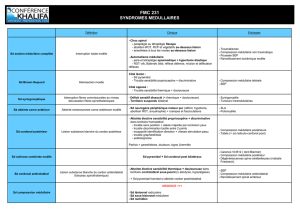
TD: Aplasie médullaire

Dr Otsmane

plan
•Définition
•Intérêts de la question
•Etude clinique
•Evolution et complication
•Formes cliniques
•DC +/ DC différentiel
•Traitement
•Conclusion

Définition
Une insuffisance médullaire quantitative IIaire à la
disparition partielle / totale de tissu hématopoïétique
Sans prolifération anormale
Congénitale ou acquise

Intérêt de la question
1) Epidémiologie:
fréquence : rare
âge: 2 pic avant 20 ans et après 50 ans
sexe : avant 50 ans pas de prédominance ,
après 50 ans =prédominance masculine.

Physiopathologie
•3 mécanismes
Lésion directe de la cell souche hématopoïétique :
- lésion acquise ( toxique chimique ou physique
- lésion congénitale : mutation modifiant la
structure de la protéine protectrice de L’ADN
Atteinte de microenvironnement : tx de soutient de
l’ hématopoïèse
Trouble auto-immunitaire : action suppressive d’une
s/population de lym T sur les cell CD34+
 6
7
8
9
10
11
12
13
14
15
16
6
7
8
9
10
11
12
13
14
15
16
1
/
16
100%