Les déficits immunitaires primitifs

Les déficits
immunitaires primitifs
Dr Messatfa Moussa
m_immuno@yahoo.fr
Faculté de médecine de Mostaganem
Cours d’immuno-pathologie
17/04/2016

I. Introduction:
Infections++++, auto-immunité, manifestations allergiques,
cancers.
La défense de l’organisme est basée sur deux
types d’immunité:
1. l’immunité innée ou non spécifique: assurée par
les barrières cutanéo-muqueuses, des facteurs
chimiques (lysozyme, acide lactique..), le
système du complément, les cellules NK et les
cellules phagocytaires (Macrophages et
polynucléaires neutrophiles).

-Ces éléments sont mis en jeu afin d’assurer :
- L ’immunité anti-infectieuse+++
-L’immunosurveillance anti tumorale
1. L’immunité spécifique: assuré par les lymphocytes et
leurs produits de sécrétion.
- Les LT:
LTCD4+: fonction helper, production de
cytokines
LTCD8+: fonction cytotoxique
- Les LB:assurent l’immunité humorale via la
production d’anticorps.

Hypersensibilité type I: anaphylaxie et atopie
En immunopathologie, plusieurs maladies ou syndromes
Sont directement tributaires à un dérèglement de ces moyens de défense
immunitaire
Système immunitaire
Auto-immunité
Déficit immunitaire
hypersensibilité
Réponse trop intense
Réponse insuffisante
ou absente
Reconnaissance du soi

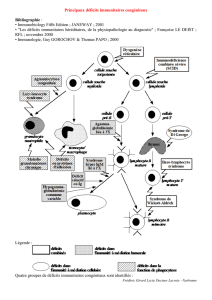
Déficits immunitaires
Primitifs (héréditaires):
Nourrisson+++
Secondaires (acquis):
À tt âge
- Déficits humoraux (B)
- Déficits cellulaires (T)
- Déficits combinés (B+T)
- Déficits de la phagocytose
- Déficit en complément
-SIDA+++
- Hémopathies malignes
- Vieillissement
- Malnutrition
- Traitement
immunosuppresseur
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
1
/
68
100%