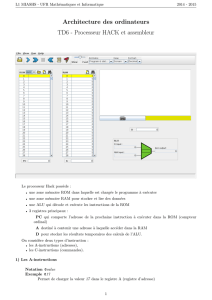
developpement clinique d`un nouveau medicament

2 avril 2004.
M-D DRICI
DEVELOPPEMENT CLINIQUE D'UN
NOUVEAU MEDICAMENT
•Quatre phases essentielles :
Phase 1, 2 et 3 (AMM) et Phase 4
•Recherche biomédicale sans ou avec
BID
•CCPPRB: avis favorable
•Consentement informé signé du
patient

2 avril 2004.
M-D DRICI
Les différents protagonistes
des Bonnes Pratiques
Cliniques
•La Loi 88.1138 du 20 décembre 1988 y fait
référence (Loi Huriet et Sérusclat).
•Le PROMOTEUR (assurance, choix inv.)
•L'INVESTIGATEUR (CCPPRB, et
consentement informé signé)
•L'ASSISTANT DE RECHERCHE
CLINIQUE

2 avril 2004.
M-D DRICI
Importance du protocole
•Introduction
•Objectif : unique
•Mesuré avec l'aide d'un
•Critère principal de jugement
•Importance du nombre de sujets
nécessaire
•Pour extrapoler les résultats de l'essai à la
population entière concernée

2 avril 2004.
M-D DRICI
Méthodologie d'essais
cliniques : Plan
1. Le développement des médicaments
Généralités, génériques et DCI
2. Les Bonnes Pratiques Cliniques
3. Rédaction d'un protocole d'essai
clinique
4. Notions de bases de méthodologie

2 avril 2004.
M-D DRICI
1. DEVELOPPEMENT
CLINIQUE D'UN NOUVEAU
MEDICAMENT
•Quatre phases essentielles
•Phase 1
•Phase 2
•Phase 3
•AMM
•Phase 4
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
1
/
74
100%