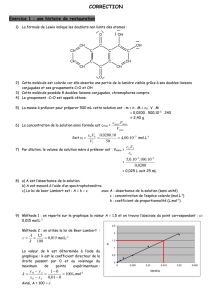
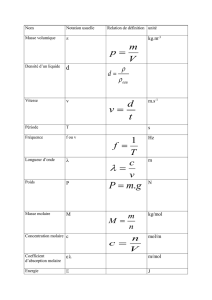
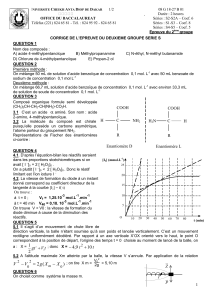
UE BCM complet

1
1
Chimie Physique
PACES 2016
UE SPE
Base Chimique du Médicament - 2016 -
Cours magistral : Josiane NURIT
UFR Pharmacie
Mardi 16/02 : Cinétique
Jeudi 18/02 : Equilibres chimiques
Jeudi 25/02 : Equilibres REDOX
Vendredi 26/02 : Equilibres REDOX
Gué Emilie UFR Pharmacie emilie-linda-marie.gue@umontpellier.fr
Nurit Josiane UFR Pharmacie josiane.nurit@umontpellier.fr
Roy Jérôme UFR Médecine jerome.roy@umontpellier.fr
Semaine du 22/02 : TD 1 : Cinétique et Équilibres
Semaine du 14/03 : TD 2 : Équilibres de solubilité et Redox
2
UE BCM
PACES 2015 - 2016
Dr Josiane NURIT - UFR Pharmacie
josiane.nurit@umontpellier.fr
© PACES UNIVERSITÉ MONTPELLIER
Chimie Physique
CINETIQUE CHIMIQUE
3
Introduction
I - Vitesse de réaction : définition
II - Loi de vitesse : ordre d’une réaction
Réactions engageant un seul réactif
A - Réaction d’ordre 0
B - Réaction d'ordre 1
C - Réaction d'ordre 2
Réactions engageant deux réactifs
D–Temps de péremption
E–Méthodes de détermination des ordres partiels
- PLAN -
III - Influence de la température sur la vitesse de réaction
A - Loi d'Arrhenius
B - Énergie d'activation
C -Calcul de l’énergie d’activation
IV - La catalyse
A - Principe
B - Les différents types de catalyse : homogène,
hétérogène, enzymatique
Conclusion

2
4
Introduction
Pour la synthèse industrielle de produits chimiques, il est fondamental
que la réaction envisagée se déroule à une vitesse suffisante
La cinétique chimique étudie la vitesse avec laquelle s’effectuent les
réactions chimiques thermodynamiquement possibles.
Certaines réactions chimiques sont instantanées, d’autres dites lentes
demandent quelques minutes, quelques heures ou quelques jours
pour amener à la formation d’un produit.
5
Introduction
Le sens des réactions
Les conditions d’équilibre
Objectifs de la thermodynamique
Alors que la Thermodynamique, grâce à des fonctions d’état
comme l’entropie et l’enthalpie libre, nous renseigne sur la faisabilité
d’une réaction.
La TD ne s’intéresse pas au chemin suivi puisque la variation des
fonctions d’état ne dépend que de l’état initial et de l’état final.
Elle permet de déterminer :
6
Introduction
La Cinétique Chimique s’intéresse au « chemin réactionnel ».
Son objectif est la mesure de la vitesse des réactions chimiques.
Objectifs de la cinétique
Un certains nombres de facteurs sont plus ou moins déterminants
dans la vitesse d’une réaction :
- La température : une élévation de température accélère
habituellement les réactions (ex les colles polymérisables durcissent
d’autant plus vite que la T est élevée)

3
7
- La concentration ou la pression partielle des
réactifs : une réaction est généralement plus
rapide qu’elles sont grandes
-Le contact entre les réactifs : les réactifs
doivent pouvoir se rencontrer et, s’ils ne sont pas
miscibles, la vitesse de réaction dépend de leurs possibilités de
contact. Dans le cas de deux liquides non miscibles, une bonne
agitation les disperse l’un dans l’autre accélérant ainsi la réaction.
- La nature du solvant
- La catalyse
- La lumière : certaines réactions ne se produisent avec une vitesse
appréciable qu’en présence de lumière (bronzage de la peau)
8
Seuls seront étudiés dans cette partie les effets de
la concentration des réactifs, de la température et
de la catalyse.
•Tout le long du chemin suivi : vitesse moyenne
•A un instant particulier : vitesse instantanée
Si l’on connaît l’équation d’une réaction et la quantité de chaque réactif
utilisé, on peut calculer la quantité de chaque produit qu’elle aura
fournie lorsqu’elle sera terminée.
On peut également déterminer le temps qui sera nécessaire pour
obtenir ces produits ou alors s’intéresser à la quantité de produit
formée pendant un intervalle de temps donné.
Notion de vitesse :
9
La notion de vitesse est très relative.
Certaines réactions sont très rapides comme :
•l’explosion des mélanges combustibles
•les réactions de dosage acido-basiques
D’autres réactions sont très lentes comme :
•la formation de la rouille
•la transformation du Cgraphiteen Cdiamant
D’autres s’effectuent à des vitesses « moyennes » :
•l’estérification des alcools
BUT :
•Augmenter ces vitesses (production
par ex)
•Diminuer ces vitesses (empêcher la formation
de rouille, permettre la conservation des
aliments)

4
10
Grâce à l’étude de la cinétique chimique nous pourrons :
Étudier les réactions de dégradation des principes actifs
Déterminer les temps de péremption des médicaments
Étudier la stabilité des principes actifs et des médicaments
Dans le domaine pharmaceutique :
11
CINETIQUE CHIMIQUE
Introduction
I - Vitesse de réaction : définition
II - Loi de vitesse : ordre d’une réaction
A - Réaction d’ordre 0
B - Réaction d'ordre 1
C - Réaction d'ordre 2
D - Temps de péremption
E –Méthodes de détermination des ordres partiels
III - Influence de la température sur la vitesse de réaction
A - Loi d'Arrhenius
B - Énergie d'activation
C - Calcul de l’énergie d’activation
IV - La catalyse
A - Principe
B - Les différents types de catalyse : homogène, hétérogène,
enzymatique
Conclusion
12
La vitesse de disparition et de formation sont
proportionnelles entre elles de la façon suivante :
aA + bB gC + dD
I - Définition de la vitesse de réaction
Disparition
des réactifs
Apparition
des produits
Soit la réaction globale :
Unité de v : mol.t-1
X = avancement de la réaction
C
A B D
dn
dn dn dn
1 1 1 1 dX
V - -
dt dt dt dt dt
a b g d

5
13
I - Définition de la vitesse de réaction
Vitesse de la réaction en fonction de l’avancement de la réaction :
Avancement d’une réaction chimique :
Lors de l’évolution de la réaction au cours du temps, les variations des
quantités de matière sont liées par les nombres stœchiométriques.
14
L'avancement d'une transformation = X
= grandeur positive exprimée en moles, qui permet de
suivre l'évolution de la composition d'un système au cours du temps.
L'état final (ou l'état d'équilibre) est atteint lorsque la composition du
système n'évolue plus.
À l'état final l'avancement Xf= avancement final.
Une transformation est dite totale si l'avancement final de la réaction
est égal à son avancement maximal. soit Xf= Xmax.
Avancement d’une réaction chimique :
15
Au cours d'une transformation totale, le réactif limitant
est entièrement consommé.
Pour déterminer la nature du réactif limitant (A), on cherche le plus
petit Xmax tel que : n(A)i-aXmax = 0 (a = coeff. stœchiométrique)
(A) est le réactif limitant, soit :
Une transformation conduit à un équilibre chimique si l'avancement
final de la réaction est inférieur à son avancement maximal,
soit Xf< Xmax
Avancement d’une réaction chimique :
ii
max
n A A V
Xaa
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
1
/
78
100%