Catalyse Enzymatique

BCM 1502 Catalyse Enzymatique Page 1/14
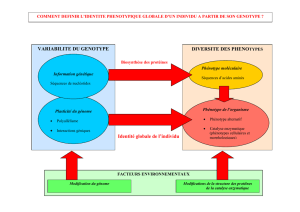
CATALYSE ENZYMATIQUE
EFFETS DE pH
La plupart des enzymes sont actifs dans une gamme de pH restreinte,
typiquement entre pH 5 à 9. Le pH influence l'activité d'un enzyme à plusieurs
niveaux :
1) par la liaison du substrat à l'enzyme
2) de l'activité catalytique
3) de l'ionisation de substrat
4) de la stabilité structurale
La vitesse initiale de beaucoup d'enzymes démontre en fonction de pH un
profil ressemblant à une cloche.
- exemple : la cinétique d’aldolase de muscle de lapin
Ces courbes reflètent l'ionisation d'un certain nombre de groupements
d’acides aminés au site actif de l'enzyme et suggèrent que certains états d'ionisation
sont essentiels pour l'expression de l'activité catalytique. Le modèle suivant est
capable d'expliquer une telle dépendance d'activité sur le pH.
1086
18
16
14
12
10
8
6
pH
Vmax (units/mg)
Curve 1
Vmax
EH + S ESH +
EP
E-ES-
EH2+ESH2+
k2k1
k-1
KE2
KE1
KES2
KES1

BCM 1502 Catalyse Enzymatique Page 2/14
Dans ce modèle simple, seulement EH et ESH possède de la compétence
catalytique. Implicite dans ce modèle, il est assumé que l'échange des protons se fait
sur le régime de l'équilibre rapide. Ceci est raisonnable vu que la constante de
deuxième ordre pour l'échange d'un proton est rapide 109 M-1.s-1. L'échange rapide
parmi les populations fait en sorte que les espèces EH et ESH sont réduites par des
facteurs, f1 et f2, dépendant de leur constante d'ionisation ou pKa. Les facteurs f1 et f2
ont la forme suivante :
]
H
[K
+ 1 +
K
]
H
[
=
f
]
H
[K
+ 1 +
K
]
H
[
=
f
+
ES2
ES1
+
2
+
E2
E1
+
1;
L'équation de Michaelis-Menten pour ce schéma possède la même forme
suivante :
] [S +
K
] [S
V
=
v
app M,
app ,
iMAX
où VMAX,app et KM,app sont des constantes michaeliennes ‘apparentes’ et
réfèrent seulement aux formes actives de l'enzyme. Cette relation ressemble à celle
obtenue de l’inhibition mixte vu que les constantes sont définis par
VMAX,app = VMAX / f2 et KM,app = KM (f1/f2).
L'analyse de la relation log VMAX,app vs pH fournie les valeurs de KES1 et KES2
correspondantes à l’état lié - tandis que la relation de log (VMAX,app / KM,app) vs pH
informe sur les valeurs de KE1 et KE2, celles de l’enzyme et substrat libre. Ceci
implique la détermination des paramètres VMAX,app et KM,app à des pH différents.
Les pKa (log KEi ou log KESi) déterminés à l’aide des logiciels donnent des
indices importants sur l'identité des acides aminés au site actif.
Par exemple, pKa ≈ 4 suggère un résidu Asp ou Glu essentiel pour l'activité de
l'enzyme. Des valeurs pKa ≈ 6 ou ≈ 10 suggère des résidus His ou Lys,
respectivement. Cependant un groupement carboxylate d'un résidu Asp en pont
salin (interaction électrostatique) avec un groupement Lys est stabilisé par la charge
positive de Lys et possède un pK plus bas (plus difficile à protoner).
NC
O
O
H
HH
pKa
Lys Asp

BCM 1502 Catalyse Enzymatique Page 3/14
Inversement un groupement aminé dans une région peu polaire est moins
basique que dans une région polaire. Dans ce cas, il s’agit du fait qu’un résidu chargé
est toujours plus difficile à stabiliser dans une région hydrophobe que sa forme
neutre correspondante.
Donc il faut être prudent dans l'identification des groupements essentiels pour
l'activité catalytique.
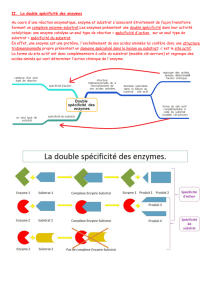
RÉACTIONS BISUBSTRATS
Les réactions enzymatiques impliquant deux substrats et deux produits
représentent 60% des réactions biochimiques connues.
Presque toutes les réactions bisubstrats sont soient des réactions transférases
où l'enzyme catalyse le transfert d'un groupement fonctionnel spécifique, X, d'un
substrat à l'autre, soient des réactions oxydation - réduction où des équivalents
réducteurs sont transférés entre deux substrats. Parmi toutes les réactions bisubstrats
(bi bi) possibles seulement quelques sont observés.
Il y a deux classifications mécanistiques majeures.
a) Réactions séquentielles ou tous les substrats doivent combiner avec
l'enzyme avant qu'il y ait transformation de substrat en produit. Le transfert du
groupe X de A (= P - X) à B donne P et Q (= B - X) est connu sous le nom de
réactions à déplacement simple. Il y a deux sortes de réactions dans l’addition des
substrats.
E
P - X + B ⇌ P + B - X
NH3
pKa
NH2Micro-environnement
hydrophobique

BCM 1502 Catalyse Enzymatique Page 4/14
i) Mécanisme séquentiel au hasard
ii) Mécanisme ordonné ou obligatoire
b) Réactions ping pong où un ou plusieurs produits sont libérés avant que
tous les substrats aient participé dans la réaction. Dans la réaction ping pong bi bi, le
produit P est libéré après l'attachement de substrat A et l'enzyme devient accepteur
du groupement X. La liaison de B à l'enzyme permet le transfert d'X au produit Q.
Mécanisme enzyme substitué (ping pong)
- ces réactions connues sous le nom déplacement double impliquent une
modification de l'enzyme et les deux substrats, A et B, ne se rencontrent pas sur la
surface de l'enzyme.
Les équations cinétiques pour le cas bisubstrats sont plus complexes et il est
possible de distinguer entre les mécanismes différents.
EA
E
Q
EAB ⇌ EPQ
EB
E
E
EP
P
P
Q
Q
A
A
B
B
A
B
P
Q
E
EA ⇌ E’P
E’
E’B ⇌ EQ
E
E
EA
EAB ⇌ EPQ
EQ
E
A
B
P
Q

BCM 1502 Catalyse Enzymatique Page 5/14
MÉCANISMES CATALYTIQUES
La catalyse est un processus qui augmente le taux par lequel une réaction
atteint l’équilibre.
La raison que les enzymes sont des catalyseurs puissants est due à leur
spécificité de liaison au substrat et leurs arrangements optimaux des groupements
catalytiques.
Les mécanismes que les enzymes utilisent sont classifiés de manière
suivante :
● L'enzyme fournit les groupes accepteur/donneur de protons, dans un
mécanisme de catalyse acide/base.
● L’enzyme peut se combiner de façon covalente avec le substrat pour former
un intermédiaire qui permet une réaction plus rapide.
● L'enzyme utilise des ions comme co-facteurs, pour stabiliser différentes
charges (intermédiaires), pour ioniser les molécules d'eau et les rendre plus
nucléophiles, et/ou pour cacher des charges.
● L'enzyme utilise les forces électrostatiques, pour aligner le substrat et
stabiliser l'état de transition.
● L'enzyme fixe le substrat de telle sorte que les liens réactionnels sont situés
près du site catalytique et sont orientés de telle sorte que l'état de transition est
atteint très rapidement.
● L'enzyme peut induire la distorsion et/ou la tension au niveau du lien à couper,
ce qui le déstabilise et le rend plus facile à couper. L'enzyme fixe donc
préférentiellement le substrat sous forme d'état de transition.
CATALYSE ACIDE/BASE
Une catalyse acide générale est un procédé dans lequel le transfert partiel d'un
proton à partir d'un groupe acide (acide Bronsted - une molécule qui peut fournir un
proton) abaisse l'énergie libre de l'état de transition.
Une réaction peut aussi être stimulée par la catalyse basique générale, si le
taux de réaction est augmenté par l'abstraction partielle d'un proton par une base
(base Bronsted - une molécule qui peut attirer un proton).
Certaines réactions utilisent les deux types de catalyse et si les deux réactions
sont simultanées, la réaction devient acide - base générale concertée.
 6
7
8
9
10
11
12
13
14
6
7
8
9
10
11
12
13
14
1
/
14
100%