Giroux-Portelance_Simon_2012_Memoire

Université de Montréal
Induction d’anticorps anti-idiotypiques contre les
protéoglycanes de la matrice extracellulaire dans la réduction
des lésions athérosclérotiques
Par
Simon Giroux Portelance
Faculté de pharmacie
Mémoire présenté à la Faculté des études supérieures et postdoctorales
en vue de l’obtention du grade de maîtrise (M.Sc.)
en sciences pharmaceutiques
option pharmacologie
Novembre, 2012
© Simon Giroux Portelance, 2012

Université de Montréal
Faculté des études supérieures et postdoctorales
Ce mémoire intitulé :
Induction d’anticorps anti-idiotypiques contre les protéoglycanes de la matrice
extracellulaire dans la réduction des lésions athérosclérotiques
Présenté par :
Simon Giroux Portelance
a été évalué par:
Sylvie Marleau, directrice de recherche
Fahima Nekka, présidente du jury
Diala Harb, membre du jury

i
Résumé
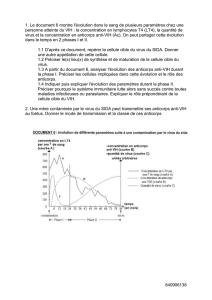
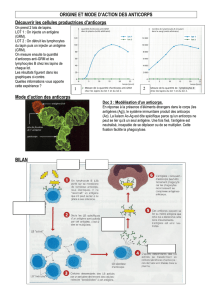
L’athérosclérose est caractérisée par l’accumulation de lipoprotéines de basse densité
(LDL) liées aux protéoglycanes de la paroi artérielle. Des anticorps chimériques (ch)
qui se lient aux glycosaminoglycanes (GAG) ont été générés. L'hypothèse est que la
vaccination avec le chP3R99, un anticorps chimérique mutant de l'hybridome P3,
pouvait interférer avec la rétention des LDL par l’induction d’une cascade d’anticorps
anti-idiotypiques dirigés contre les GAG.
Des souris mâles déficientes en apolipoprotéine E ont été soumises à une diète
hypercholestérolémique et ont reçu 5 injections sous-cutanées de 50 µg de vaccin
chP3R99 ou de vaccin chP3S98 (un mutant de faible réactivité). Les injections ont été
effectuées à chaque semaine ou aux 2 semaines. Au moment du sacrifice, l'aorte
perfusée avec du PBS a été excisée et analysée après coloration au Oil Red-O. Les
résultats ont été exprimés en pourcentage de lésions sur la superficie totale de l'aorte.
La réactivité contre le chP3R99, chP3S98, l’héparine, le sulfate de dermatane et de
chondroïtine des sérums de souris immunisées a été mesurée par ELISA. De plus, la
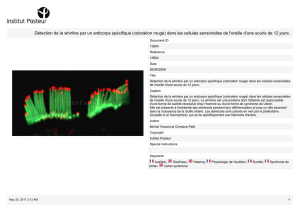
liaison de l'anticorps chP3R99 aux GAG dans la lésion d'athérosclérose a été observée
par un appareil de visualisation in vivo.

ii
Nos résultats montrent que l’immunogénicité des anticorps chP3R99 est supérieure à
celle des anticorps chP3S98 et que le sérum des souris immunisées avec le chP3R99
présente des anticorps anti-idiotypiques dirigés contre les GAG. Cet effet est associé à
une réduction de 42 % (p < 0.01) du pourcentage de lésions athérosclérotiques chez les
souris vaccinées.
L'utilisation d’une immunisation active avec l’anticorps chP3R99 pourrait constituer
une approche thérapeutique pour le traitement de l'athérosclérose.
Mots-clés : Vaccin, anticorps, chP3R99 , athérosclérose, cascade anti-idiotypique,
glycosaminoglycane, souris déficientes en apolipoprotéine E.

iii
Abstract
Atherosclerosis is characterized by the accumulation of low density lipoprotein (LDL)
associated with the proteoglycans of the arterial wall. Chimeric (ch) antibodies that
react against glycosaminoglycans (GAG) were generated. We tested the hypothesis
that vaccination with chP3R99, a mutant chimeric antibody of the P3 hybridoma, could
interfere with the retention of LDL by inducing a cascade of anti-idiotypic antibodies
directed against the GAG.
Male mice deficient in apolipoprotein E fed with a hypercholesterolemic diet were
given five subcutaneous injections of 50 µg of chP3R99 or chP3S98 (a mutant with
low reactivity) vaccine. The injections were performed every week or every two
weeks. After sacrifice, the aorta was perfused with PBS, excised and analyzed after
staining with Oil Red-O. The results were expressed as a percentage of lesions areas on
the total area of the aorta. The reactivity of the sera obtained was tested against the
chP3R99, chP3S98, heparin, dermatan and chondroïtin sulfate from obtained
immunized mice by ELISA. The anti-idiotypic response was measured by blocking the
anti-isotypic reactivity by a nonspecific IgG, hR3. In addition, the antibody chP3R99
binding to GAG in the atherosclerotic lesion was shown by an in vivo molecular
imaging device.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
1
/
106
100%