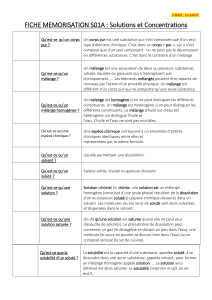
1.1. Réactions acide-base

1. Réactions Acides - Bases

1.1. Définitions
•Dans les réactions « acide-base »:
•Particule échangée :
–Proton H+
•Noyau de l’atome d’hydrogène
•La demi-réaction acide-base fondamentale, d’après la théorie
de BRÖNSTED est :
Donneur ↔ Accepteur + H+
–Donneur de proton = acide
–Accepteur de proton = base
–couple (Donneur / Accepteur) = couple acide-base
conjugués

1.1. Définitions
•Exemple :
CH3COOH
↔
CH3COO-+H+
Donneur1

1.1. Définitions
•Exemple :
CH3COOH
↔
CH3COO-+H+
Donneur1 Accepteur1

1.1. Définitions
•Exemple :
CH3COOH
↔
CH3COO-+H+
Donneur1 Accepteur1
ACIDE1
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
1
/
73
100%