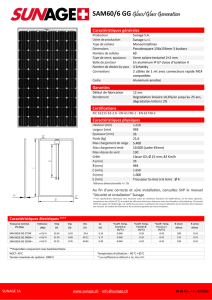
Graphique 1 - Faculté de Médecine et de Pharmacie Fès

UNIVERSITESIDIMOHAMMEDBENABDELLAH
FACULTEDEMEDECINEETDEPHARMACIE
FES
AnnéeThèseN°/201009010
UNIVERSITESIDIMOHAMMED
BENABDELLAH
FES
SYNDROMED’ACTIVATIONMACROPHAGIQUE
CHEZL’ENFANT
(Aproposde12cas)
THESE
PRESENTEEETSOUTENUEPUBLIQUEMENTLE10/05/2010
PAR
Néele06Janvier1984àNador
Mlle.AMARAKAOUTAR
POURL'OBTENTIONDUDOCTORATENMEDECINE
MOTS-CLES:
Enfant-Activationmacrophagique-Fièvre-Splénomégalie
Leishmanioseviscérale
JURY
M. Professeur
M.
Professeur
.Professeur
Mme.
Professeur
HIDAMOUSTAPHA........................................................
dePédiatrie
me.........................................................
agrégéde
M............................................................
agrégéde
.......................................................
agrégéde
BONOWAFAA Médecineinterne
ATMANISAMIR Pédiatrie
CHAOUKISANA Pédiatrie
JUGE
PRESIDENTETRAPPORTEUR

1
LISTE DES ABREVIATIONS
ACJ : Arthrite chronique juvénile
ADP : Adénopathie
AEG : Altération de l’état général
BAAR : bacilles acido-alcolo résistants
BAHS : Syndrome hémophagocytaire associé aux bactéries
BOM : Biopsie ostéomédullaire
CCMH : Concentration corpusculaire moyenne en hémoglobine
CFU-G : Colony Forming Unit-Granulocytic
CFU-GM : Colony Forming Unit Granulo-Monocyt
CFU-M : Colony Forming Unit-Monocytic
CG : Culot globulaire
CHS : Syndrome de Chediak –Higashi
CIVD : Coagulation intra-vasculaire disséminée
CMV : cytomégalovirus
CP : Culot plaquettaire
CRP : C reactiv protein
CSF : Colony stimulating factor
CVC : Circulation veineuse collatérale
EBV : Epstein barr virus
ECBU : Examen cytobactériologique des urines
EHb : Electrophorèse d’hémoglobine
EPP : Electrophorèse des protides
GB : Globules blancs
GGT : Gamma Glutamyl Transférase
Hb : Hémoglobine
HHV : human herpes virus
HLA : humain leucocyt antigen
HSV : herpes simplex virus
HTLV : Human T cell lymphotropic virus
HVA : Virus de l’hépatite A
HVB : Virus de l’hépatite B
HVC : Virus de l’hépatite C
IDR : Intradermoréaction
IFN : Interféron
IL : Interleukine
LCR : Liquide céphalo-rachidien
LDH : Lactates deshydrogénase
LH : Lymphohistiocytose

2
LHF : Lymphohistiocytose hémophagocytaire familiale
LVI : Leishmaniose viscérale infantile
LYST : LYSosomal Traffıcking regulator
M-CSF : Monocytic-Colony Stimulating Factor
MNI : Mononucléose infectieuse
MNP : système mononuclé phagocytaire
MQ : Macrophages
NFS : Numération formule sanguine
NK : Cellules natural killer
NSE : Estérase non spécifique
PAL : Phosphatase alcaline
Pâleur CM : Pâleur cutanéo-muqueuse
PFC : Plasma frais congelé
PL : Ponction lombaire
PNN : Polynucléaires neutrophiles
RMH : Réticulose médullaire histiocytaire
SAM : Syndrome d’activation macrophagique
SAP : SLAM Associated Protein
sCD25 (=sIL-2R) : récepteur soluble de l’interleukine2
SGOT : Sérum GlutamoOxaloacetate Transférase
SGPT : Sérum GlutamoPyruvate Transférase
SLAM : Signaling Lymphocytic Activation Molecule
SMG : Splénomégalie
SNC : Système nerveux central
TCA : temps de céphaline active
TDM : Tomodensitométrie
TG : Triglycérides
Th : lymphocytes T helper
TNF : Tumor necrosis factor
TP : Taux de prothrombine
VAHS : Virus associated hemophagocytic syndrome
VGM : Volume globulaire moyen
VIH : Virus de l’immunodéficience humaine
VLDL : Very Low Density Lipoprotein
VS : Vitesse de sédimentation
VZV : Virus varicelle zona

3
SOMMAIRE
INTRODUCTION ............................................................................................. 6
DEFINITION ................................................................................................... 09
HISTORIQUE .................................................................................................. 11
PHYSIOPATHOLOGIE ....................................................................................... 15
I. Généralités sur les macrophages ...................................................................... 16
II. Mécanismes du SAM....................................................................................... 25
III. Conséquences de l'activation macrophagique ................................................. 31
PATIENTS ET METHODES ................................................................................ 35
I. Buts de notre étude.......................................................................................... 36
II. Matériel d’étude .............................................................................................. 36
III. Méthodes d’étude .......................................................................................... 36
IV. Critères d’inclusion ........................................................................................ 36
V. Nos observations ............................................................................................ 43
RESULTATS.................................................................................................... 94
I. Données épidémiologiques .............................................................................. 95
II. Etude clinique ................................................................................................. 98
III. Etude paraclinique ........................................................................................ 102
IV. Critères diagnostiques .................................................................................. 108
V. Etiologies ...................................................................................................... 109
VI. Traitement .................................................................................................... 110
VII. Evolution ..................................................................................................... 111
DISCUSSION ................................................................................................. 112
I. Epidémiologie ................................................................................................. 113
II. Diagnostic positif ........................................................................................... 116

4
1. Présentation clinique .............................................................................. 116
A.Signes généraux .................................................................................. 117
B. Organomégalie ................................................................................... 117
C. Signes cutanés ................................................................................... 117
D. Signes neurolgiques ........................................................................... 118
E. Syndrome oedémato-ascitique ............................................................ 119
F. Signes pulmonaires ............................................................................. 120
G. Signes digestifs .................................................................................. 120
H. Autres manifestations cliniques .......................................................... 120
2. Signes biologiques ................................................................................. 123
A. Anomalies de l’hémogramme.............................................................. 124
B. Troubles del’hémostase ...................................................................... 125
C. Atteinte hépatique .............................................................................. 126
D. Bilan lipidique .................................................................................... 126
E. Hyperfirritinémie ................................................................................. 127
F. Troubles hydroéléctrolytiques ............................................................. 127
G. Autres anomalies biologiques ............................................................. 128
3. Signes cyto-histologiques ...................................................................... 131
A. Cytologie ............................................................................................ 131
B. Histologie ........................................................................................... 135
4. Critères dignostiques ............................................................................. 138
III. Diagnostic différentiel ................................................................................... 142
IV. Etiologies...................................................................................................... 143
1. Les SAM primaires .................................................................................. 144
2. Les SAM secondaires .............................................................................. 155
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
1
/
227
100%