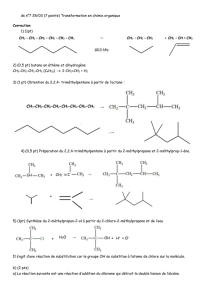
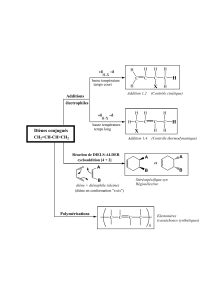
Croissance de la chaîne polymérique

Chimie organique
Les matériaux polymères organiques
Mécanismes d’obtention
I Généralités sur les polymères - définitions :
1. Définitions :
a. Notion de polymère :
• Un polymère est une macromolécule, de masse molaire élevée, obtenue par la répétition
d’un motif constitutif.
Exemples :
Le polyéthylène est le polymère constitué de la répétition du motif constitutif éthylène
(-CH2-CH2-). Le polymère est représenté par la formule {;—CH2-CH2 }n;— , et est
synthétisé au laboratoire. Voici quelques autres exemples :
On peut trouver des polymères naturels, comme la cellulose.
• La polymérisation est une réaction qui conduit à un polymère par enchaînement de
motifs. Les molécules qui s'enchaînent sont appelées monomères.
Polyéthylène PE
Polypropylène PP
Polychlorure de vinyle
Polytétrafluoroéthylène
Polystyrène
Polymétacrylate de méthyle
PVC
PTFE
PS
PMMA

Le PE a été obtenu par réactions successives de beaucoup de molécules d’éthylène
CH2=CH2.
• Si on a fait s’enchaîner des monomères de nature différente, on a fabriqué un copolymère.
b. Structure macromoléculaire des polymères :
Les polymères ne sont pas constitués de molécules strictement identiques. Il ne s’agit pas
d’un corps pur ! On verra dans la suite du cours que, selon leur mode de synthèse, on peut
avoir des topologies très différentes :
• Polymère linéaire : les chaînes sont linéaires, mais n’ont pas toutes la même longueur.
• Polymère ramifié : des branches secondaires sont liées à la chaîne principale.
• Polymère réticulé : les chaînes sont liées entre elles.
On verra que cette multitude de structure possible fait que les propriétés macroscopiques
peuvent être très différentes d’un polymère à l’autre.
c. Polymolécularité d’un polymère :

Comme le polymère est constitué de molécules qui ne sont pas toutes identiques, on est
obligé de définir des grandeurs moyennes sur le polymère.
• Distribution en masse molaire :
Soit une chaîne correspondant à la suite de i motifs constitutifs.
Ce nombre est noté DPi : dégré de polymérisation
Dans le polymère, on trouve Ni moles de molécules de degré de polymérisation DPi.
Notons M0 la masse molaire du motif constitutif.
La masse molaire de la chaîne caractérisée par DPi vaut Mi = DPi.M0
On peut tracer la distribution en masse molaire, qui est Ni = f(Mi)
Avec des masses molaires élevées, la distribution est « continue » :
Ces figures illustrent bien le fait que le polymère n’est pas un corps pur.
• Masses molaires moyennes d’un polymère :

On définit la masse molaire moyenne en nombre par , xi étant la fraction
molaire de la chaîne de degré DPi. On a alors, puisque :
Masse molaire moyenne en nombre d’un polymère :
On définit la masse molaire moyenne en masse par , wi étant la fraction
massique de la chaîne de degré DPi. On a alors, puisque :
Masse molaire moyenne d’un polymère :
• Indice de polymolécularité :
Par définition, l’indice de polymolécularité, Ip , est donné par :
On peut montrer qu’en notant l’écart type sur la distribution en masse molaire, on a
. Ainsi, il est clair que :
Ip > 1.
Ip = 1 pour un corps constitué de molécules identiques.
Ip est d’autant plus élévé que la dispersion en masse molaire est grande
Mnxi.Mi
i1
xiNi
Nj
j1
Mn
Ni.Mi
i1
Ni
i1
Mwwi.Mi
i1
wimi
mj
j1
Ni.Mi
Nj.Mj
j1
Mw
Ni.Mi
2
i1
Ni.Mi
i1
IPMw
Mn
IP12
Mn
2

Démonstration :
L’écart type est défini par le fait que, pour une distribution x, 2 est égal à la différence
entre la moyenne des x2 et du carré de la moyenne des x : 2 = moy(x2) - (moy(x))2. Ainsi,
pour l’écart-type sur la distribution en masse molaire, on a :
2 =
d’où :
2. Différents types de polymérisation :
On distingue deux grands types de polymérisation (trois, avec la polymérisation par les
métallocènes, type réaction de Ziegler-Natta, hors programme …).
a. La polymérisation par étapes :
Dans la polymérisation par étapes, des molécules ayant au moins deux groupes
fonctionnels se soudent les unes aux autres, par réactions chimiques (estérification,
formation d’amides, etc …)
Ni.Mi
2
i1
Ni
i1
Ni.Mi
i1
Ni
i1
2
Ni
i1
.Ni.Mi
2
i1
Ni
i1
2
Ni.Mi
i1
Ni
i1
2
Ni
i1
.Ni.Mi
2
i1
Ni.Mi
i1
2.
Ni.Mi
i1
2
Ni
i1
2
Ni.Mi
i1
Ni
i1
2
Ni.Mi
i1
Ni
i1
2
.
Ni
i1
.Ni.Mi
2
i1
Ni.Mi
i1
21
Mn
2.Ip1
IP12
Mn
2
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
1
/
21
100%