TP de culture cellulaire

TP de culture cellulaire
I. Introduction
Aujourd’hui, l’embryologie est une science qui s’est tournée vers la compréhension des
mécanismes qui permettent le développement des organismes. Pour la plupart des oiseaux, et
notamment pour notre poule, le développement de l’embryon se fait en dehors de l’organisme
maternel, à l’abri d’une protection, vulgairement appelée œuf.
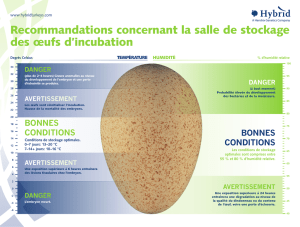
Cet œuf contient toutes les ressources nécessaires au développement de l’embryon, que ce soit
au niveau de l’énergie, de l’eau et de la protection. L’enveloppe externe de cet embryon
permet donc la protection de l’embryon, de par sa composition en 3 parties principales :
Vitellus, Amnios et Allantoïde. Nous verrons les rôles de ces enveloppes dans la première
partie.
En ce qui concerne le développement de l’embryon en lui-même, un certain nombre de
processus se mettent en place, que ce soit pour la division cellulaire en elle-même, ou pour la
reconnaissance, la migration et la différenciation cellulaire. Ces mécanismes ont pour finalité
de former les tissus et les organes de la future poule.
Composant ces tissus et organes, un grand nombre de molécules sont aujourd’hui connues,
aussi bien au niveau de la matrice extracellulaire qu’au niveau des membranes. Nous pouvons
de plus, à ce jour, mettre en culture des cellules, et ainsi mieux comprendre les mécanismes
biologiques et cellulaires. Pour notre TP, nous voulons mettre 2 types de cellules en culture,
des cellules cérébrales et des fibroblastes. Pour les mettre en culture, nous utilisons la
technique de culture de cellules adhérentes à un support.Nos observations se font au
microscope inversé, sous hotte à flux laminaire, et donc en conditions stériles.
II. 1ère partie : mécanisme d’adhésion et de migration cellulaire
A. L’embryon et les annexes embryonnaires
Figure 1 : Schéma de l'embryon de poulet à 7 jours de développement
Figure 2 : Position relative de l'Embryon dans l'œuf, en vue du dessus, en ayant coupé
du côté du "gros bout"

Figure 3 : Photo de la position de l'œuf par rapport aux annexes embryonnaires une fois
l’embryon extrait de l’œuf.
Figure 4:Zone cérébrale d'un embryon d'œuf de poule
B. Rôle des annexes
a) Vitellus
Réserves nutritives de l’embryon, rôle nourricier.
b) Vésicule vitelline
Entoure la masse de Vitellus, sert d’organe digestif. Elle est parsemée de vaisseaux sanguins,
ainsi les produits sont absorbés par ceux-ci et transportés vers l’embryon.
c) Allantoïde :
Fonction dans la respiration : la splanchnopleure amène une vascularisation ce qui permet des
échanges gazeux entre l’allanto-chorion et la coquille de l’œuf.
Fonction nutritive sur plusieurs aspects :prend le relais du raphé séro-amniotique pour
l’assimilation du reste de l’albumen et permet de récupérer le calcium de la coquille pour
former les os de l’embryonce quifragilise la coquille et ainsi facilite la sortie du poussin.
Fonction excrétrice : la cavité allantoïde est une poubelle de l’embryon, en effet les déchets
produits par les reins seront lâchés dans l’allantoïde. Au moment de l’éclosion l’allanto-
chorion reste collée à la coquille.
d) Chorion
Couche extra-embryonnaire externe constituée de mésoderme et d’ectoderme.
e) Cœlome extra-embryonnaire
Cavité générale de l’œuf.
f) Amnios
Permet le développement de l’embryon en milieu liquide.Permet l’assimilation de l’albumen
par le raphé séro-amniotique jusqu'à 16 jours d’incubation.Assure à l’embryon une protection
contre une possible dessiccation et d’éventuels chocs mécaniques.
C. Mise en culture des cellules cérébrales embryonnaires de poulet
Matériel et méthodes :
1. Milieu DMEM
a) DMEM base
Le milieu DMEM base est acheté tel quel.

b) DMEM 5%
Le milieu DMEM 20% se prépare en suivant la composition suivante :
- Milieu de base : DMEM 500 mL
- Sérum de veau fœtal (SVF) 25 mL (5%)
- Antibiotique 6 mL
- L-glutamine 6 mL
c) DMEM 20%
Le milieu DMEM 5% se prépare en suivant la composition suivante :
- Milieu de base : DMEM 500 mL
- Sérum de beau fœtal (SVF) 110 mL (20%)
- Antibiotique 6 mL
- L-glutamine 6 mL
2. Poly Lysine
Pour 10 boites :
1- Peser l’acide borique dans un bécher de 50 mL et ajouter 40 mL environ d’H2O
2- Ensuite, peser la poly lysine dans un micro bécher à l’aide de 2 pinces et l’ajouter avec
attention à l’acide borique. Agiter le tout.
3- Peser le NaOH 1N dans un bécher de 25mL et ajouter de l’H2O puis mélanger avec
une tige de verre dans un bain-marie. Ensuite, le transvaser dans une fiole et ajuster à
50 mL. Agiter et mettre dans un flacon.
4- Le PH étant de 4,75 environ, il faut (0,5mL environ d’NaOH 1N) pour remonter le pH
à 8,4
5- Ensuite ajuster la solution à 50 mL avec H2O
6- Prévoir un bécher de 25 ou 50 mL et une seringue de 10 mL avec un filtre de 0,22 µm
et verser 2,5mL par boite de Pétri (sous la hotte). Ne plus bouger les boites jusqu’à la
mise en culture.
3. Tampon borate
Pour les boites « borate », la procédure est la même sauf que l’on n’ajoute pas la poly lysine
au tampon borate.
4. Fibronectine
On veut préparer 10 boites de 60 mm de diamètre, ayant donc chacune 28 cm² de surface. On
veut 2 µg.cm-2, on a donc besoin de 560 µg de fibronectine.
Pour cela on suit le protocole suivant :
1- Reconstituer avec 1 mL d’eau stérile par mg de protéine. Laisser dissoudre pendant au
moins 30 minutes.
2- Diluer la fibronectine dans une solution saline tamponnée (la dilution n’est pas
indiquée) et recouvrir la surface de culture avec un volume minimum.
3- Laisser sécher à l’air pendant au moins 45 minutes à température ambiante. L’excès de
fibronectine sera éliminé par aspiration.
Lors de notre manipulation, nous avons donc mis en culture des cellules cérébrales
embryonnaires de poulet dans des boites de Pétri présentant différents revêtements afin de
savoir lequel semblait être le plus adapté à ce type de culture cellulaire.
Protocole:

Répartir 1,5 mL de la solution cellulaire + 3,5 mL de DMEM 20% dans chaque boite
D. Analyse et interprétation des résultats
1. Tampon borate
Après 1 heure d’incubation à 37°C en incubateur à CO2, on observe une absence d’adhérence
des cellules sur le fond de la boite. On observe la présence d’amas cellulaires, sans doute dus
à une mauvaise séparation des cellules lors de la manipulation.
Figure 5 : Culture de neurones en tampon borate après 1 heure d'incubation
Après 4 jours d’incubation, les cellules n’ont toujours pas adhéré, on constate l’absence de
croissance cellulaire, et la présence de quelques corps apoptotiques (cellules mortes).
Après 7 jours d’incubation, les cellules n’ont ni adhéré, ni poussé, et seuls les corps
apoptotiques sont visibles.
Figure 6 : Culture de neurones en tampon borate après 4 jours d'incubation

Figure 7 : Culture de neurones en tampon borate après 7 jours d'incubation
Donc, le tampon borate n’est pas un milieu approprié pour la croissance descellules cérébrales
embryonnaires, en effet, dans les conditions expérimentales, nous n’avons observé aucune
adhérence, migration ou croissance de celles-ci.
2. Polylysine
Après 1 heure d’incubation à 37°C en incubateur à CO2, on observe l’apparition de petits
prolongements cellulaires sur certaines cellules, ceci montre que les cellules sont entrain
d’adhérer à la paroi de la boite. Par ailleurs, on peut observer l’apparition de longs
prolongements qui pourraient traduire un début de croissance cellulaire.
Figure 8: Culture de cellules cérébrales embryonnaires sur polylysineaprès 1h d'incubation.
Après 4 jours d’incubation, on constate la présence de corps apoptotiques. Les cellules
vivantes adhèrent à la paroi. De plus, les cellules ont poussé, de nombreux prolongements
(axones et dendrites) sont apparus. On remarque aussi les synapses entre différents neurones.
Figure 9 : Culture de cellules cérébrales embryonnaires sur polylysine après 4 jours d'incubation.
Figure 10 : Culture de cellules cérébrales embryonnaires sur polylysine après 7 jours d'incubation.
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%