Document

Eléments de microscopie optique et
électronique
Marc Landry
UFR SDV, Université Bordeaux Segalen
IINS, CNRS UMR 5297

Les échelles de grandeur
Microscope de Hooke (XVIIème siècle)

1. Bases de la microscopie
1.1. Dualité onde-corpuscule
1.2. Eléments d’un microscope
1.3. Formation d’une image en microscopie optique
1.4. Electron et rayonnement

1.1. Dualité onde-corpuscule
• Fréquence: n = c/l
• Energie: E = hn
•Ondes : diffraction, interférences
•Corpuscules : collision
De Broglie

1.2. Eléments d’un microscope
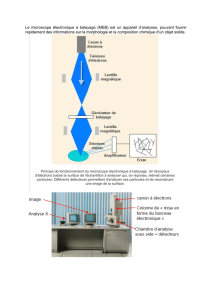
Deux catégories de microscopes
Tout microscope est constitué :
•d’une source de rayonnement
•d’un échantillon
•d’un détecteur de la vitesse et des directions des particules après collision
•Détecteur loin de l’échantillon: microscopie de champ lointain
•Pas de séparation entre source, échantillon et détecteur (sonde
locale): microscopie de champ proche
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
1
/
80
100%