memoire de magister

MINISTERE DE L’ENSEIGNEMENT SUPERIEUR ET DE LA RECHERCHE SCIENTIFIQUE
UNIVERSITE MOULOUD MAMMERI DE TIZI-OUZOU
FACULTE DES SCIENCES- DEPARTEMENT DE PHYSIQUE
MEMOIRE DE MAGISTER
SPECIALITE : PHYSIQUE
OPTION : SCIENCE DE LA MATIERE
Présenté par :
HADDAG OUIZA
Sujet : « Etude par dynamique moléculaire des propriétés de transport
atomique et des excitations collectives dans les métaux liquides »
Devant le jury d’examen composé de :
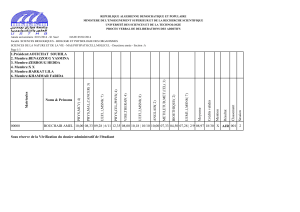
TIGRINE Rachid Professeur UMMTO Président
HELLAL Slimane Professeur UMMTO Rapporteur
LALAM Fadila Professeur UMMTO Examinatrice
BOURAHLA Boualem Maître de Conférences(A) UMMTO Examinateur
HARCHAOUI Nadra Maître de Conférences(B) UMMTO Examinatrice
Soutenu le : 13/ 12 / 2012

Remerciements
Dans ce mémoire, nous rapportons l’essentiel de notre travail de recherche sur les
propriétés physiques des métaux alcalino-terreux à l’état liquide. Il a été réalisé à l’université
Mouloud Mammeri de Tizi-Ouzou (UMMTO) au Laboratoire de Physique et Chimie
Quantique (LPCQ), sous la direction scientifique du Professeur Hellal Slimane. Je lui suis très
reconnaissante de m’avoir initiée à un thème pour le moins difficile mais combien captivant :
la physique de l’état liquide. Grâce à lui, j’ai été familiarisée avec les principaux outils
d’étude théorique de la matière dense désordonnée. On peut citer : la méthode du pseudo-
potentiel, la simulation numérique et plus spécialement les fonctions de corrélations. Je ne
saurais oublier de lui témoigner mon entière satisfaction quant à ses qualités humaines. Ainsi
son art d’aborder et d’expliquer avec clarté les aspects les plus difficiles mais cruciaux de la
théorie m’a grandement facilité la tâche. Ses conseils, ses encouragements toujours
renouvelés et son entière disponibilité sont également le reflet d’un profond humanisme. Qu’il
en soit vivement remercié.
J’exprime ma vive reconnaissance à monsieur el hadj Tigrine Rachid, Professeur à la
faculté des Sciences de l’UMMTO, qui m’a fait l’honneur de bien vouloir assurer la
présidence du jury de ce mémoire. Je veux associer dans un même remerciement Madame
Lalam Fadhila, Professeur, Madame Harchaoui Nadra, Maître de conférences et Monsieur
Bourahla Boualem, Maître de conférences. Ceux-ci, enseignants à l’UMMTO, ont bien voulu
examiner le manuscrit et de figurer dans mon jury.
J’exprime ici mon sincère remerciement à Monsieur Bouarab Said, Professeur
l’UMMTO, qui en tant que directeur du LPCQ, m’a assuré les conditions de travail des plus
satisfaisantes au laboratoire. Je renouvelle aussi l’expression de ma profonde amitié à
l’adresse de Melle Azzaz Dihia, secrétaire du Laboratoire, pour sa gentillesse et son
dévouement. Elle a ainsi toujours répondu favorablement à toutes mes sollicitations.
Ma gratitude va à tous les membres du laboratoire qui ont tous plus ou moins
contribué à l’avancement de mes travaux ne fût-ce que par l’expression de leur amitié à mon
égard.
Je remercie également toutes celles et tous ceux auprès desquels j’ai obtenu aide,
conseils et soutien moral pour la réalisation de ce mémoire. Je pense notamment à tous les
membres de ma famille et à tous mes proches.
Que les personnes oubliées dans ces remerciements d’avance me pardonnent !

A mes très chers parents
A tous les êtres qui me sont chers

« Le Seigneur est subtil, mais Il n’est pas malveillant ».
Albert Einstein

i
Table des matières
Introduction générale 1
Chapitre I
De l’Hamiltonien mono-électronique aux potentiels effectifs interioniques dans
les métaux
I.1 Introduction 4
I.2 Energie totale dans un métal dans l’approximation non relativiste 4
I.3 Equation mono-électronique et approximations fondamentales 5
I.4 Méthode du pseudopotentiel 7
I.5 Ecrantage auto-cohérent : fonction diélectrique 11
I.6 Développement au deuxième ordre en série de perturbation de l’énergie électronique 13
I.7 Energie totale dans un métal : potentiel effectif interionique 14
I.8 Classes de pseudo-potentiels modèles : pseudopotentiels ab initio BHS et OB 17
I.9 Application du formalisme : potentiels effectifs interioniques des alcalino-terreux 23
I.10 Conclusion du chapitre I 25
Figures du chapitre I 26
Bibliographie du chapitre I 31
Chapitre II
Du potentiel effectif aux propriétés statiques et dynamiques des métaux liquides
II.1 Fondements de la dynamique moléculaire 34
II.2 Introduction aux propriétés structurales 38
II.3 Introduction aux propriétés de transport atomique 45
II.4 Corrélations spatio-temporelles 51
II.5 Conclusion au chapitre II 54
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
1
/
85
100%