Les différentes maladies du coeur - Institut Saint

Les différentes maladies du coeur
Auteur : Dr Pascal AMEDRO
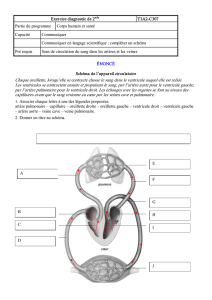
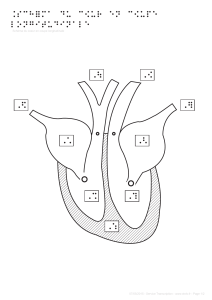
1. Le cœur normal
L’oxygène capté dans l’air va dans les poumons, où le sang «bleu» est alors oxygéné et
devient «rouge». Il est conduit par les 4 veines pulmonaires jusqu’à l’oreillette gauche, qui
charge le «ventricule gauche», pour qu’il éjecte le sang à forte pression dans l’aorte. Tous
les organes utilisent alors ce sang oxygéné, et une fois redevenu «bleu», il retourne au
coeur par les 2 veines caves jusqu’à l’oreillette droite. Celle-ci charge le ventricule droit qui
va éjecter le sang bleu à basse pression dans les artères pulmonaires. Ainsi la boucle est
bouclée et on reprend un nouveau cycle.
Quatre valves s’ouvrent et se ferment en fonction du cycle cardiaque : la valve aortique,
entre le ventricule gauche et l’aorte; la valve mitrale entre l’oreillette gauche et le ventricule
gauche; la valve pulmonaire entre le ventricule droit et l’artère pulmonaire.
Le coeur est sous la dépendance d’une conduction électrique, qui part du noeud sinusal
(dans l’oreillette droite), emprunte des faisceaux électriques vers le noeud auriculo-
ventriculaire jusqu’aux deux ventricules. Le rythme cardiaque est de l’ordre de 80 à 180
battements par minutes chez un nouveau-né ; il baisse progressivement jusqu’à 60 à 80
battements /minutes chez l’adulte.
http://www.dailymotion.com/swf/video/x1aajb

2.
Principales Malformations cardiaques
Presque un enfant sur 100 nait avec une malformation cardiaque, cela représente la
première cause d’anomalies congénitales. On parle aussi de «cardiopathies
congénitales». Plus de la moitié sont relativement «bénignes», autorisant une vie normale
après chirurgie cardiaque ou cathétérisme interventionnel. Environ 20% de ces
malformations cardiaques sont considérées comme complexes et doivent être prises en
charge depuis la période foetale jusqu’à l’âge adulte par des médecins spécialistes en
cardiologie congénitale.
Communication Inter-Auriculaire (CIA) : un trou, de taille variable, est présent entre les
deux oreillettes. En général, les enfants n’ont aucun symptôme et on découvre cette
anomalie après avoir entendu un souffle cardiaque au stéthoscope. La CIA peut soit se
fermer spontanément, soit persister. Dans la majorité des cas, il sera fermé par le
cardiopédiatre au cours d’un cathétérisme cardiaque interventionnel : une prothèse est
mise en place sous anesthésie générale et l’enfant sort de l’hôpital le lendemain
(http://www.amplatzer.com/products/asd_devices/the_amplatzer_septal_occluder/tabid/18
8/default.aspx). Si le cathétérisme n’est pas possible en raison de l’anatomie de la CIA, le
patient sera confié au chirurgien cardiaque qui fermera le trou par une suture ou un patch.
Comme une CIA existe de façon normale chez le foetus, le diagnostic anténatal est
rarement possible.

Communication Inter-Ventriculaire (CIV) : un trou, de taille variable, est présent entre
les deux ventricules. S’il est petit, l’enfant vit normalement, n’a aucun symptôme et est
régulièrement surveillé par le cardiopédiatre. Si la CIV est importante, le nourrisson grossit
mal, s’essouffle lorsqu’il tête et nécessite souvent un traitement médicamenteux. On parle
alors d’insuffisance cardiaque. Si la CIV ne se ferme pas spontanément ou que
l’insuffisance cardiaque ne se stabilise pas, une fermeture par chirurgie cardiaque est
nécessaire. Le diagnostic anténatal des CIV larges est possible.
Persistance du Canal Artériel (PCA) : le canal artériel est un tuyau qui existe
physiologiquement chez le foetus, entre l’artère pulmonaire et l’aorte. Il se ferme
normalement après la naissance. S’il persiste et qu’il est significatif, l’enfant risque de se
mettre en insuffisance cardiaque. Le canal artériel devra être fermé par le cardiopédiatre
au cours d’un cathétérisme interventionnel. L’enfant quitte l’hôpital en général le
lendemain du cathétérisme. Rarement, l’enfant est adressé au chirurgien cardiaque qui
ligature le canal artériel.

Coarctation de l’Aorte : lors de la fermeture du canal artériel après la naissance, il arrive
que l’aorte se ferme ou se resserre également. Le nouveau-né risque alors un malaise
grave, le nourrisson une insuffisance cardiaque, l’enfant et le jeune adulte une
hypertension artérielle, l’adulte un accident vasculaire cérébral. Chez les plus jeunes, le
traitement est la chirurgie cardiaque ; chez les autres, on effectue un cathétérisme
cardiaque interventionnel. Le diagnostic anténatal de cette pathologie est rarement
possible sauf si l’anomalie est associée à d’autres malformations du coeur.
Tétralogie de Fallot : il s’agit d’une anomalie embryologique qui a quatre conséquences
anatomiques: une CIV, une aorte déviée à droite, un obstacle entre le ventricule droit et
l’artère pulmonaire et un ventricule droit «musclé». Le diagnostic anténatal est souvent
réalisé. A la naissance, le nouveau-né risque des accès de «cyanose», où il devient bleu.
Un traitement médicamenteux peut être nécessaire, voire la mise en place par le
chirurgien cardiaque d’un tuyau qui remplace le canal artériel («Blalock»). Le plus souvent,
vers l’âge de 6 mois, la cure complète est effectuée par le chirurgien : fermeture de la CIV,
ouverture de l’obstacle entre le ventricule droit et l’artère pulmonaire.

Atrésie Pulmonaire avec CIV (APSO) : c’est la forme extrême de Tétralogie de Fallot, ici
le sang ne passe pas du tout entre le ventricule droit et l’artère pulmonaire. Ici la chirurgie
à la naissance est nécessaire, avec mise en place d’un tuyau entre l’aorte et l’artère
pulmonaire («Blalock»). La réparation par chirurgie cardiaque est plus complexe mais a
lieu en général vers l’âge de 6 mois. Il s’agit d’une cardiopathie congénitale complexe
devant être prise en charge par un cardiopédiatre puis par un cardiologue congénitaliste à
l’âge adulte.
Canal atrio-ventriculaire : il s’agit d’un trou situé dans la région centrale de division du
coeur, entre les oreillettes et les ventricules; il existe plusieurs formes isolées ou
associées : CIA, CIV, fente sur la valve mitrale. Dans les formes complètes, parfois
associées à une anomalie génétique (trisomie 21), le nourrisson est en insuffisance
cardiaque et le traitement est chirurgical. Le diagnostic anténatal est souvent possible.
Transposition des gros vaisseaux : l’aorte et l’artère pulmonaire sont connectées de
façon inversée aux deux ventricules. Le diagnostic est le plus souvent fait pendant la
grossesse, même si elle ne gêne pas le foetus. Le nouveau-né doit naître dans une
maternité de niveau III disposant d’un service de réanimation néonatale et d’une astreinte
de cardiopédiatrie 24h/24h. A la naissance le bébé devient immédiatement «bleu», un
cathétérisme est effectué dans la couveuse par le cardiopédiatre pour élargir la CIA et
parfois un médicament gardant le canal artériel ouvert est nécessaire. Après quelques
jours de vie, l’intervention chirurgicale peu avoir lieu, c’est le «switch artériel», qui remet en
place les deux gros vaisseaux ainsi que les artères coronaires. Il s’agit d’une cardiopathie
congénitale complexe devant être prise en charge par un cardiopédiatre puis par un
cardiologue congénitaliste à l’âge adulte.
 6
7
8
6
7
8
1
/
8
100%