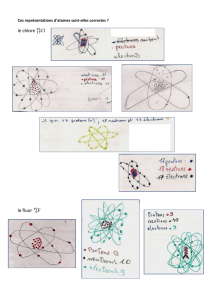
Chimie 1 : Structure de la matière - E

Polycopié de cours :
Structure de la matière
Présenté par :
Fatiha BARKA-BOUAIFEL
2014-2015

AVANT-PROPOS
Le présent polycopié de cours que je présente, dans le cadre de mon habilitation (HDR)
-communs Sciences de la matière (SM),
ral
de chimie mais également à tous ceux qui doivent connaitre les bases modernes de cette
science, sans pour autant devoir en traiter chaque jour en spécialiste.
Il porte essentiellement sur les notions fondamentales de chimie générale (structure de la
m
négligeables. Pour cela, un rappel de quelques notions fondamentales (les états de la matière
et méthodes de séparation, les atomes et molécules, les solutions) est nécessaire pour la
compréhension du programme que ce soit en cours, en travaux dirigés ou en travaux
pratiques.
Les notions les plus modernes dans le domaine de la structure de la matière ont été plus
détaillées dans ce cours.
les isotopes, réactions nucléaires,
-atomique (dualité
onde-
spectre des ions hydrogénoïdes, modèle de Sommerfeld).
Le chapitre I
(
Schr
Le chapitre IV est dédié à la classification périodique des éléments où seront traités plusieurs
points (principe de la classification périodique, lois et propriétés, propriétés physiques et
chimiques des familles d'éléments, les familles chimiques).
Le dernier chapitre est réservé à la liaison chimique (la liaison ionique, la liaison covalente,
structure de Lewis, la méthode VSEPR, la liaison covalente dans le modèle quantique,
hybridation des orbitales atomiques, la liaison polarisée, la liaison métallique, les liaisons
intermoléculaires).
recrutement en 1997 à ce jour au sein de plusieurs départements à
(

SOMMAIRE
Rappels sur les notions fondamentales ............................................................... 1
Chapitre I ........................................................................ 8
Chapitre II -atomique) .................... 18
Chapitre III ...................................................... 29
Chapitre IV : Classification périodique des éléments ....................................... 45
Chapitre V : La liaison chimique ...................................................................... 56
Références bibliographiques ............................................................................ 94

1
Rappels sur les notions fondamentales
La matière est constituée de particules élémentaires : les atomes. Actuellement, il y a 114
La matière se trouve sous forme de mélanges (homogène ou hétérogène) de corps purs. Un
corps pur est caractérisé par ses propriétés physiques (température de fusion, température
deux
catégories de corps purs :
Corps purs simples ent (exemple : O2, O3, H2,
Corps purs composés constitués de deux ou plusieurs éléments (exemple :
H2O, FeCl2, HCl, H2SO4,
I. Etats de la matièreet méthodes de séparation
La matière existe sous trois formes : solide, liquide et gaz. Le froid et la chaleur jouent un rôle
très
liquide se fait
. Ces transformations sont illustrées par la figure 1 ci-dessous :
Figure 1. Les états de la matière et ses transformations.
Il existe plusieurs techniques de séparation
et des constituants de corps purs composés en corps purs simples
(électrolyse, radiolyse, etc).
I.1.Méthodes de séparation des corps purs composés
e séparer les corps purs composés et comprend les procédés
qui permettent de décomposer un corps composé.
Exemple :

2
Les procédés utilisés dans ce cas sont de nature chimique. On cite particulièrement :
La thermolyse ou pyrolyse qui permet
chaleur.
La radiolyse qui consiste à décomposer une substance par des radiations lumineuses
(UV, visibles ou IR).
qui permet
I.2.Méthodes de séparation des constituants d’un mélange
en corps purs. Cette
séparation est basée sur les différences des propriétés physiques de ces constituants.
1. Mélanges hétérogènes
Si le mélange est constitué de plusieurs solides, on utilise : le tamisage (différence de
taille des particules), la lévigation (différence de masse volumique) comme dans le cas
de la
sable, la dissolution
Si le mélange est constitué de solides et de liquides, on utilise la filtration ou la
centrifugation.
Si le mélange est constitué de liquides, on utilise la décantation, comme dans le cas
du mélange eau-huile.
2. Mélanges homogènes
Dans ce cas, on peut utiliser la distillation fractionnée (différence de tempéra
des liquides), la précipitation(cristallis),
Exemple : Séparation par précipitationuncationen milieu acide à
, notamment par spectrophotométrie de masse, permet de dles
éléments présents dans le composé et de déterminer leurs proportions.
Exemple composé de 78% N2et 22% O2. Les deux gaz
.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
1
/
97
100%