Chimie du Benzène: Substitution Électrophile Aromatique

CHAPITRE 16 : CHIMIE DU BENZENE : SUBSTITUTION ELECTROPHILE SUR LES AROMATIQUES
5
Chapitre 16 : Chimie du Benzène : Substitution électrophile sur les aromatiques
16.0.Mécanisme général 35-36
Etape 1 : Attaque électrophile
E+E
H
E
H
E
H
Carbocation cyclohexadiènyle
Il faut utiliser un électrophile fort puissant pour qu’il puisse attaquer le noyau
aromatique.
Etape 2 : perte d’un proton
E
H
E
H
E
HE
+ H
+
Diagramme d’énergie potentielle
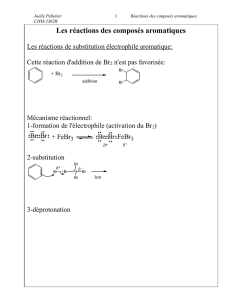
16.1.Bromation des noyaux aromatiques 38-41
+ Br
2
FeBr3 + HBr
B
r

CHAPITRE 16 : CHIMIE DU BENZENE : SUBSTITUTION ELECTROPHILE SUR LES AROMATIQUES
6
Il faut impérativement utiliser un catalyseur (FeBr3, AlCl3 (acides de Lewis)) afin
d’augmenter le caractère électrophile de Br2 (sans catalyseur, il n’est pas assez électrophile
pour s’attaquer au noyau aromatique).
Action du catalyseur :
Mécanisme : cf. mécanisme général (16.0.)
N.B. Au labo, on va générer le catalyseur in situ en mettant un clou en fer par
exemple qui va générer des Fe3+.
16.2.Chloration et iodation des aromatiques 42-52
Chloration :
Attention : ne pas utiliser FeBr3 comme catalyseur sinon il pourrais y avoir des
échanges avec les Cl-.
Catalyseur : AlCl3, FeCl3.
+ Cl
2
FeCl
3
+ HCl
Cl
Iodation :
Cette réaction est très lente. Pour la catalyser on va utiliser de l’eau oxygénée et du
Cu+2 (en fait ce ne seront plus des catalyseurs mais presque des réactifs a part entière car on
va devoir les employés en « grande » quantité).
I
2
+ 2 Cu
+2
2 "I
+
" + 2 Cu
+
Mécanisme : cf mécanisme général (E+= I+) et le H sera arracher par I-).
On peux aussi utiliser I2 avec AgNO3, ce qui créera le complexe acide base de Lewis
Ag+NO3- et en fin de réaction formera du AgI qui précipite.
Nitration des aromatiques :
Cette réaction est très lente. Il va falloir activer HNO3 grâce à l’acide sulfurique.
HO N
O
O
HOSO
3HN
O
O
+ HSO4-
HO
H
H2O + ONO
ion nitronium
Mécanisme : cf. mécanisme général (HSO4- ou H2O joue le rôle de base). Comme
c’est NO2+ qui produit l’attaque, on pourrait aussi utiliser du NO2+BF4- ou NO2+PF6-.
La nitration va permettre de synthétiser des diisocyanates qui servent ensuite a faire
des polymère (polyuréthane).
En plus d’un rôle de catalyseur, l’acide va permettre de dessécher le milieu. Il a
cependant quelques inconvénients. Le principal est la pollution qu’il cause, on tente donc
de plus en plus à le remplacer avec des zéolithes par exemple.
Réduction des composés nitrés (H2, Fe, SnCl2,…)

CHAPITRE 16 : CHIMIE DU BENZENE : SUBSTITUTION ELECTROPHILE SUR LES AROMATIQUES
7
NO
2
NH
2
1. SnCl
2
,H
3
O
+
2. OH
-
Le traitement basique permet de déprotonner l’amine.
Sulfonation :
SO
3
H
H
2
SO
4
/ SO
3
(8%)
Mécanisme : cf. mécanisme général (attaque de SO3 ou +SO3H).
Cette réaction est quasiment thermoneutre, on a donc un processus équilibré et on
peux très bien désulfoné (les conditions sont un peu plus dures).
SO
3
H
H
2
O, H
2
SO
4
(catalyseur) H
100°C
+ HOSO
3
H
Dans des condition plus dure encore (NaOH, 300°C), on peux pratiquer une fusion
sodique de ces acides benzosulfonique (on change le –SO3H en –OH)
On va pouvoir aussi convertir (après déprotonation de l’acide) les acidesylfonique en
chlorure de sulfonyle en les faisant réagir avec du SOCl2 ou PCl5.
S
O
O
ONa+
2+ PCl
5
SO2Cl
+ POCl3 + 2NaCl
SO
2
Cl
OH
+N(Et)
3
OSO
2
H
3
C
CH
3
On se place en milieu basique afin d’éviter la formation de HCl.
On utilise aussi ces chlorures de sulfonyle pour synthétiser des sulfamides (qui sont des
composés très important en pharmacie).
Ar SO
2
Cl + NHR
2
ArSO
2
NR
2
+ HCl
16.3. Alkylation des noyaux aromatiques (réaction de Friedel Craft) 53-56
Schéma général :
+ RX AlX3+ HX
R
H
Réactivité de l’halogénoalcane : RI < RBr < RCl < RF
Réactivité de l’acide de Lewis : BF3 < SbCl5 < FeCl3 < AlCl3 < AlBr3
Cette réaction pose quand même quelques problèmes lorsqu’on utilise des dérivés
fort peu encombré, en effet, l’alkyl va enrichir le cycle en électron et donc le rendre plus
réactif, on aura donc des produits de polyalkylation (avec un dérivé tertiaires,
l’encombrement stérique empêche une seconde attaque).

CHAPITRE 16 : CHIMIE DU BENZENE : SUBSTITUTION ELECTROPHILE SUR LES AROMATIQUES
8
Mécanisme : cf. mécanisme général de l’addition électrophile sur aromatiques (X—
AlX3 va déprotoner).
Action du catalyseur
Il y a différentes extensions possibles à la réaction de Friedel-Crafts.
Alkylation de Friedel-Crafts intramoléculaire
On va pouvoir aussi utiliser d’autres précurseurs de carbocations comme les alcool ou
encore un alcène.
Alcools :
H
H
3
CCH
2
C
H
OH
CH
3
+BF
3,
60°C, 9h
H
CCH
2
CH
3
H
3
C
(CH3)3COH + SbF5
SO2 (solvant) CH3C
CH3
CH3
+ HOSbF5
Alcènes :
H
2
CCHCH
2
H
+
H
3
CH
CCH
3
Limitations de la réaction de Friedel-Crafts :
1. Les halogénures d’aryle ou de vinyle ne sont pas réactifs.
2. Les noyaux aromatiques désactivés par un groupement électroattracteurs ne donnent pas
de réactions.
N.B. : en général les groupements –NH2, --NHR et --NR2 sont des groupements activant,
mais dans ce cas non, en effet, l’acide de Lewis va attaquer la paire libre de l’azote et donc
la rendre électroattractrice.
3. Polyalkylation : les groupements alkyls sont électrodonneurs par effets inductifs, ils
enrichissent donc le cycle aromatique (des R—X suffisament encombré permettent de limiter
les dégâts).
4. Les carbocations peuvent se réarranger et donc diminuer le rendement en produits
attendu ainsi que de produire un mélange de produit assez difficile à séparer. Les
carbocations peuvent se réarranger de plusieurs manières : migration d’hydrure, migration
de CH3-.

CHAPITRE 16 : CHIMIE DU BENZENE : SUBSTITUTION ELECTROPHILE SUR LES AROMATIQUES
9
16.4. Acylation ou alcanoylation de Friedel-Crafts 57-62
H
C
O
ClH
3
C
+
AlCl
3
CCH
3
O
L’astuce qui permet cette réaction est la
formation d’un ion acylium :
CORRCO
Pour cela, on va tout d’abord faire réagir un halogénure d’alcanoyle ou un
anhydride carboxylique avec un acide de Lewis pour former un complexe qui sera un
précurseur d’acylium.
La réaction est similaire pour un anhydrine (R—C(O)—O—AlCl3 est le groupe partant).
On terminera toujours la réaction par un traitement aqueux car AlCl3 peut encore
réagir avec le produit de cette manière :
C
RR'
O
+AlCl
3
C
RR'
O
A
lCl
3
3H
2
OC
RR'
O
+ AlOH
3
+ 3HCl
L’avantage principal de cette réaction est que le groupe acyle va désactiver le
noyau aromatique et on n’aura donc pas de produits de polyacylation.
Il existe une extension de cette réaction : la réaction de Gatterman-Koch.
CH
3
H
+ CO + HCl AlCl
3
, CuCl
CH
3
CHO
Dans les 2 cas, on pourra réduire la fonction carbonylée afin de retrouver un alkyl
(réduction de Clemmensen en milieu acide, réaction de Wolff Kishner en milieu basique et
formation de thioacétal / réduction avec du Ni de Raney en milieu neutre).
16.5.Réactivité des noyaux aromatiques 62-66
On va faire la différence entre 2 types de substituants :
 6
7
8
9
10
11
6
7
8
9
10
11
1
/
11
100%