Malformations corticales et épilepsie : Apport de l`IRM

Malformations corticales et
épilepsie : Apport de l’IRM
JP Cottier, C Sembely, M Bosc, S Gallas ,
C Vinikoff-Sonier S, D Lo, D Herbreteau ,
D Sirinelli
CHU TOURS

INTRODUCTION
•Malformations corticales reconnues
comme fréquentes causes d’ épilepsie
- Avant IRM: 4% des épilepsies
intraitables médicalement
- Avec IRM: 11à 40 % des séries

Intérêt du dépistage et de la
classification de ces malformations
1) L’ épilepsie devient souvent réfractaire au traitement
médical et un traitement chirurgical est envisagé
- L’ IRM précise la nature et topographie de la lésion
- La possibilité de la chirurgie
- L’ évolution probable après chirurgie
2) La Base génétique des MC est mieux comprise
Le conseil génétique constitue une part importante de l’
assistance aux familles

But du travail:
Présenter l’ aspect IRM de ces
malformations corticales épileptogènes
en y associant leurs aspects cliniques,
génétiques et thérapeutiques

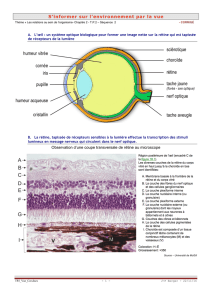
Les trois événements fondamentaux de la formation
corticale : base pour une classification des
malformations
1) Prolifération cellulaire
A partir de la 7 SA, prolifération et
différentiation dans les régions
périventriculaires
2) Migration neuronale
Préférentiellement le long des
prolongements radiaires des cellules
gliales
3) Organisation corticale
Longue période de différentiation et de formation synaptique
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1
/
57
100%