transparents 1ere partie.

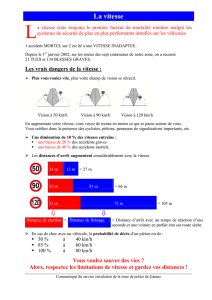
Introduction à la
Phylogénie moléculaire
Abdoulaye Baniré Diallo
(Université du Québec à Montréal)
Plan
!Introduction
!Les données moléculaires
!Rappel sur les arbres
!Les méthodes d’inférence phylogénétique
•Les méthodes de distances
•Les méthodes de parcimonie
•Les méthodes de Maximum de vraisemblance
•Les méthodes Bayésiennes
!Les validations statistiques
!Splits and Spectra
!Les réseaux phylogénétiques
!Les phylogénies connues
!Simulation
!Sujets divers
Aujourd’hui-Semaine 1
!Introduction à l’analyse phylogénétique
•Le problème et les applications
!Les données moléculaires
•Les caractères et les distances
!Rappel sur les arbres
•Arbre enraciné vs arbre non enraciné
•Arbres binaires
•Nombre de topologies possibles
•Dénombrement de topologies
!Les méthodes de distances
•Principe
•Relation matrice-phylogénie
•Algorithmes PGM
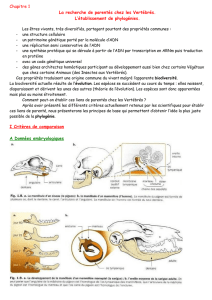
Arbre et relations inter-espèces

Arbre et relations inter-espèces
!Darwin (1859) a initié l’arbre comme support formel de la
représentation des relations inter-espèces
!Au début les modes de classifications des espèces étaient:
•Les comparaisons morphologiques
•Les comparaisons comportementales
•Les répartitions géographiques
!Aujourd’hui les phylogénies obtenues à partir:
•des séquences moléculaires (phylogénie moléculaire)
•des caractères discrets
•des fréquences des gènes
•des traits quantitatifs
•des sites de restriction
•des microsatellites
Phylogénie-phylogenèse
!Définition:
•l’histoire de la formation et de l’évolution
d’une espèce, d’un phylum (série
évolutive des formes animales dérivant
d’un ancêtre commun)
!Du grec
•Phûlon = tribus
•Genesis = origine
Arbre et relations inter-espèces
!l'histoire du
développement
paléontologique des
organismes par
analogie avec
l'ontogénie ou histoire
du développement
individuel
!Haeckel (1860)
Arbre et relations inter-espèces

Plus de 3000 articles traitent
de l’analyse phylogénétique
La phylogénie moléculaire
!Définition:
•La phylogénie moléculaire est la discipline ayant
pour objectif la reconstruction de l'histoire
évolutive des espèces par comparaison des
séquences de leurs gènes ou de leurs protéines.
!Données:
•Un ensemble d’organismes (taxa) et pour chacun
un ensemble de données moléculaires
(séquences par exemple).
La phylogénie moléculaire
(Données)
Taxons Caractères
Espèce A ATGGCTATTCTTATAGTACG
Espèce B ATCGCTAGTCTTATATTACA
Espèce C TTCACTAGACCTGTGGTCCA
Espèce D TTGACCAGACCTGTGGTCCG
Espèce E TTGACCAGTTCTCTAGTTCG
La phylogénie moléculaire
(Résultats)
Taxon B
L’espace entre les taxons et leur
position (en terme de hauteur)
ne signifie rien.
Taxon A
Taxon C
Taxon E
Taxon D
Cette dimension peut avoir des longueurs
de branches identiques (cladogramme et ultramétrique)
ou non identiques (arbre additif ou phylogramme)

La phylogénie moléculaire
Applications
!Projet: Tree of life
•Avec plus de 4000
pages web, le projet
présente la diversité des
organismes sur la terre,
leurs histoires
évolutionaires et leurs
caractéristiques
Histoire de l’évolution
Évolution des caractères
La phylogénie moléculaire
Applications
!Bio-écologie
•Déplacement d’espèces
•Relation hôtes-parasites

La phylogénie moléculaire
Applications
!Épidémiologie
•Tracer l’évolution d’un
virus à travers ces
différentes souches
(dentiste)
Utilisation de la phylogénie pour comprendre
les phénomènes de duplications et pertes de
gènes
A. Arbre de gène B. L’arbre de gènes superposé à un
arbre d’espèces pour identifier
les pertes de gènes.
Est ce que le Dr David Acer a contaminé ses patients ?
DENTIST
DENTIST
Patient D
Patient F
Patient C
Patient A
Patient G
Patient B
Patient E
Patient A
Local control 2
Local control 3
Local control 9
Local control 35
Local control 3
Oui:
Les séquences de VIH de 5
de ses patients sont dans le
même clade que les
séquences de VIH du Dr
Acer.
No
No
Ou et al. (1992), Page et Holmes (1998)
Arbre phylogénétique
des séquences de VIH
du DENTISTE, ses
7 patients et
35 infectés
dans la même région
Géographique.
Est ce que le Dr Richard Smith a contaminé sa
femme?
!En 1998, la femme du Dr Richard accuse son
mari médecin de l’avoir délibérément injecté du
sang contaminé au VIH.
!Des arbres d’évolutions du virus ont prouvé que
le médecin a effectivement contaminé sa
femme.
!Premières preuves d’arbres d’évolutions
acceptés par une cour criminelle aux USA.
!Il a été condamné à 50 ans de prison pour
meurtre au second degré.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
1
/
33
100%