MEMOIRE - Université des Sciences et de la Technologie d`Oran

R
épublique
A
lgérienne
D
émocratique et
P
opulaire
M
inistère de l’
E
nseignement
S
upérieur et de la
R
echerche
S
cientifique
U
niversité des
S
ciences et de la
T
echnologie d’
O
ran
M
ohamed
B
oudiaf
Faculté de Mathématiques et Informatique Département d’Informatique
MEMOIRE
En vue de l’obtention du
Diplôme de Magistère
Le jury est composé de :
Pr
Mr. BENYETTOU Abdelkader
Pr Mme
. IZABATENE FIZAZI Hadria
Pr Mr. BENYETTOU Mohamed
Dr Mr. BELKADI Khaled
Président
Examinatrice
Rapporteur
Examinateur
USTO-MB
USTO-MB
USTO-MB
USTO-MB
Année Universitaire 2015 / 2016
Présenté et Soutenu par :
Mr. MOUSSATI Omar
Intitulé
Classification des données de biopuces
Domaine : Mathématiques et Informatique.
Spécialité : Informatique.
Intitulé de la Formation : Simulation et Intelligence Artificielle.

Abstract Medical diagnosis is very important in the field of recognition and
treatment of disease. The biochip is a modern technology which helps us to make the
diagnosis. To treat our subject of classification, we presented the various stages of an analysis
by DNA chip, then we have applied selection methods such as Fisher test and the BW test on
selecting population. The KNN algorithm and the dual explorations genetic algorithm are
applied to all the genes selected for classification. The classification rate we found show that
our results are competitive. We plan to do a combination of different methods of selection and
to validate the results biologically.
Keywords— Biochips ; Microarray; Data Classification ; DNA chip .
Résumé Le diagnostic médical est un élément très important dans le domaine de
reconnaissance et traitement des maladies. La biopuce est l’une des techniques modernes qui
nous aide à faire le diagnostic. Pour traiter notre sujet de classification, nous avons présenté
les différentes étapes d’une analyse par puce à ADN, puis nous avons appliqué des méthodes
de sélection telles que le test de Fisher et le test BW sur la population de sélection.
L’algorithme Kppv et l’algorithme génétique à double explorations sont appliqués sur
l’ensemble des gènes sélectionnés pour faire la classification. Les taux de classification que
nous avons trouvés montrent que nos résultats sont compétitifs. Nous envisageons de faire
une combinaison entre les différentes méthodes de sélection et de valider les résultats
biologiquement.
Mots-clés : Biopuces; Classification des données; Puce à ADN.

I
Dédicaces
Je dédie ce travail
A Mes très chers parents qui m’ont aidé et encouragé durant toutes les années
de mes études.
A Tous mes enseignants.
A Mes frères et sœurs.
A Toute la famille MOUSSATI et A Tous mes amis.

II
Remerciements
Je remercie DIEU qui m’a guidé dans mes pas et qui m’a donné le courage tout au long du
développement de ce mémoire.
Je tiens à exprimer mes sentiments de gratitude et de reconnaissance à mon encadreur
Monsieur Mohamed BENYETTOU, professeur à l’université Des sciences et technologie
d’Oran (USTO) , qui a consacré son temps à suivre de près l’évolution de ce projet, à
orienter les différentes étapes et à pallier toutes les difficultés auxquelles j’ai eu à faire face.
Je souhaiterais vivement remercier l’ensemble de l’équipe de Simulation et Intelligence
Artificielle, je tiens aussi à remercier tous les étudiants de S.I.A et M. O. E.P.S, pour
avoir créé une ambiance chaleureuse et amicale tout au long de mes études.
Je suis aussi reconnaissant envers tous les enseignants qui ont contribué durant toutes
mes études. Ainsi, tous ceux qui m’ont aidé de près ou de loin à réaliser ce modeste travail.
J’adresse ma reconnaissance à tous les personnes qui mon fait l’honneur d’avoir
participé à mon jury.

Table des matières
III
Table des matières
Introduction ...............................................................................................................................2
Chapitre1 : Notions Biologiques Elémentaires.
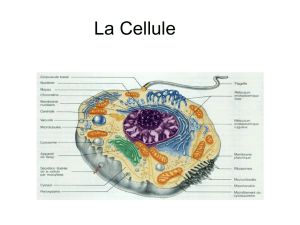
1.1 La cellule .............................................................................................................................6
1.2 Acide désoxyribonucléique (ADN) …………………………….……………..………….6
1.3 Le transcriptome…………… …………………………………………………..….……...7
1.4 La méthode P C R .……………………………………………………….…….………....8
1.4.1 Introduction .………………………………… …………………….…………….…8
1.4.2 Principe de la PCR :……………………………………………….………..……….9
1.5 Historique des biopuces :………………………………………………....…….…….….10
1.6 Synthèse du chapitre :………………………………………………………………..….13
Chapitre2 : Principe des biopuces.
2.1 Principe des puces à ADN ………………………………….………………..…….........16
2.1.1 La préparation des cibles et l’hybridation ………………….…..……..…….…..17
2.1.2 Acquisition et analyse des images …………………… ……..…………..…........17
2.1.3 Transformation des données ………………………………………….……........18
2.2. Plateformes :………………………………………………………….…………..……..20
2.2.1 Technologie Agilent …………………………………………………..……........20
2.2.1.1 Avantages ………………………………………………..…………..….20
2.2.1.2 Inconvénients …………………………………………………….…. ....21
2.2.2 Technologie Affymetrix :…………………………………………….……… …..21
2.2.2.1 Avantages . ………………………………………………………… …...21
2.2.2.2 Inconvénients………………………………………………………… …22
2.3 Domaines d’application ………………………………………….…..………………… 22
2.3.1 L’environnement ………………………………………….…………………... ...22
2.3.2 Diagnostics médicaux ………………………………………………..………... ..23
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
1
/
78
100%