Trouble du rythme et de la conduction chez l`enfant - chu

Journal
de
pédiatrie
et
de
puériculture
(2016)
29,
191—210
Disponible
en
ligne
sur
ScienceDirect
www.sciencedirect.com
ARTICLE
EMC
Trouble
du
rythme
et
de
la
conduction
chez
l’enfant夽
Arrhythmia
and
conduction
in
children
A.
Maltreta,∗,b
aCentre
de
référence
des
malformations
cardiaques
congénitales
complexes
(M3C),
hôpital
Necker—Enfants-Malades,
149,
rue
de
Sèvres,
75015
Paris,
France
bInstitut
cardiovasculaire
Paris
Sud,
6,
avenue
Noyer-Lambert,
91300
Massy,
France
MOTS
CLÉS
Tachycardie
;
Bradycardie
;
Rythme
irrégulier
;
Tachycardie
supraventriculaire
;
Cardiomyopathie
rythmique
;
Trouble
du
rythme
héréditaire
;
Traitement
antiarythmique
;
Bloc
auriculo-
ventriculaire
;
Ablation
endocavitaire
Résumé
La
grande
majorité
des
troubles
du
rythme
à
l’âge
pédiatrique
est
représentée
par
des
tachycardies
supraventriculaires.
L’évolution
naturelle
des
tachycardies
de
la
première
année
de
vie
est
favorable.
La
prise
en
charge
consiste
en
un
traitement
antiarythmique
de
quelques
mois
pour
éviter
les
récidives
et
écarter
le
risque
de
cardiomyopathie
rythmique.
Au-delà
de
5
à
10
ans,
un
traitement
curatif
par
ablation
endocavitaire
peut
être
envisagé.
Les
troubles
conductifs
de
l’enfant
sont
observés
dans
deux
contextes
distincts.
Dans
la
période
périnatale,
il
s’agit
presque
exclusivement
de
bloc
auriculo-ventriculaire
complet
d’emblée
d’origine
immunologique.
Les
diagnostics
de
trouble
conductif
fait
au-delà
de
cette
période
sont
plus
volontiers
évolutifs,
se
présentant
initialement
comme
des
troubles
conductifs
partiels.
La
stimulation
cardiaque
permanente
est
indiquée
en
cas
de
bradycardie
symptomatique
ou
de
fréquence
cardiaque
inférieure
à
50
battements
par
minute.
Les
troubles
du
rythme
héréditaires
ou
canalopathies
sont
rares
mais
potentiellement
létaux.
Leur
prise
en
charge
doit
se
faire
dans
un
centre
spécialisé.
La
prévention
de
la
mort
subite
dans
ce
contexte
passe
par
des
modifications
du
mode
de
vie,
un
traitement
médical
et/ou
l’implantation
d’un
stimulateur
cardiaque
ou
d’un
défibrillateur
automatique
implantable.
©
2016
Publi´
e
par
Elsevier
Masson
SAS.
夽Grâce
au
partenariat
mis
en
place
en
2010
entre
le
Journal
de
Pédiatrie
et
de
Puériculture
et
l’EMC,
les
articles
de
cette
rubrique
sont
issus
des
traités
EMC.
Celui-ci
porte
la
mention
suivante
:
A.
Maltret.
Trouble
du
rythme
et
de
la
conduction
chez
l’enfant.
EMC
—
Pédiatrie
-
cardiologie
2015
[Article
4-107-A-70].
Nous
remercions
l’auteur
qui
a
accepté
que
son
texte,
publié
initialement
dans
les
traités
EM,
puisse
être
repris
ici.
∗Correspondance.
Adresse
e-mail
:
http://dx.doi.org/10.1016/j.jpp.2016.06.002
0987-7983/©
2016
Publi´
e
par
Elsevier
Masson
SAS.

192
A.
Maltret
Introduction
Les
mécanismes
des
arythmies
de
l’enfant,
sans
malforma-
tion
cardiaque
associée,
sont
les
mêmes
que
chez
l’adulte
mais
leur
épidémiologie
et
leur
histoire
naturelle
sont
singu-
lières.
La
pédiatrie
est,
comme
chacun
sait,
une
spécialité
d’âge
et
non
d’organe.
Cet
adage
se
vérifie
pour
les
troubles
du
rythme
et
de
la
conduction.
Durant
l’enfance,
la
présen-
tation
clinique,
les
diagnostics
à
évoquer,
la
prise
en
charge
et
l’évolution
d’une
arythmie
ou
d’un
trouble
conductif
sont
différents
selon
que
l’enfant
a
quelques
jours
de
vie
ou
plusieurs
années.
Cette
prise
en
charge
ne
peut
s’envisager
sans
un
diagnostic
précis.
L’électrocardiogramme
(ECG)
est
le
principal
outil
diagnostique,
c’est
pourquoi
cet
article
s’articule
autour
des
différents
aspects
électrocar-
diographiques
en
tachycardie,
bradycardie
ou
rythme
irrégulier.
Tachycardie
régulière
à
QRS
fins
La
très
grande
majorité
des
arythmies
de
l’enfance
est
représentée
par
des
tachycardies
régulières
à
QRS
fins
soit,
par
définition,
des
tachycardies
supraventriculaires
(TSV).
Le
terme
TSV
regroupe
différentes
formes
d’arythmies
de
localisation
et
de
mécanisme
différents.
Par
définition,
ce
sont
des
arythmies
qui
naissent
en
amont
de
la
bifurca-
tion
du
faisceau
de
His.
Le
QRS
est
fin
(moins
de
80
ms).
La
cadence
ventriculaire
dépasse
souvent
230
battements
par
minute
(bpm)
chez
le
nouveau-né.
La
présentation
cli-
nique
est
fonction
de
l’âge
de
l’enfant
et
de
la
tolérance
hémodynamique
de
la
tachycardie.
Dans
la
première
année
de
vie,
le
diagnostic
peut
être
fortuit
à
l’auscultation
lors
d’un
examen
systématique.
À
l’inverse,
l’entrée
dans
la
maladie
peut
être
dramatique
dans
un
tableau
de
col-
lapsus
circulatoire
si
une
tachycardie
persistante
a
été
méconnue
jusqu’à
se
compliquer
d’une
cardiomyopathie
rythmique.
L’association
avec
une
cardiopathie
congénitale
n’est
pas
rare
:
10
à
15
%
[1],
voire
plus
en
cas
de
syndrome
de
Wolff—Parkinson—White
[2].
Le
diagnostic
du
type
de
TSV
repose
sur
l’analyse
du
rapport
entre
les
activités
atriales
(onde
P
ou
P)
et
ventriculaires
(QRS).
La
distinction
sur
l’ECG
de
sur-
face
de
ces
événements
n’est
pas
toujours
aisée
chez
l’enfant,
non
seulement
parce
que
le
cycle
de
la
tachy-
cardie
est
rapide
mais
aussi
parce
qu’elle
est
conduite
en
1/1.
Les
manœuvres
vagales,
voire
l’enregistrement
œsophagien,
sont
souvent
utiles
et
peuvent
éventuelle-
ment
être
couplés.
Les
manœuvres
vagales
chez
l’enfant
sont
:
l’application
d’une
vessie
de
glace
sur
le
visage
pendant
10
secondes
chez
le
nouveau-né
et
la
compres-
sion
des
globes
oculaires
chez
les
plus
grands.
Le
test
à
l’adénosine
est
souvent
nécessaire
(adénosine
avant
1
an
:
0,15
mg/kg,
après
1
an
:
0,1
à
0,3
mg/kg
en
intraveineuse
flash
:
Krenosin®—Adénocor®;
l’adénosine
triphosphate
[Striadyne®]
:
0,5
mg/kg
en
intraveineuse
flash).
Tachycardie
jonctionnelle
par
réentrée
Tachycardie
jonctionnelle
réciproque
par
une
voie
accessoire
La
tachycardie
jonctionnelle
réciproque
ou
rythme
réci-
proque
orthodromique
est
une
réentrée
qui
descend
par
le
nœud
auriculo-ventriculaire
puis
par
le
His
et
remonte
par
une
voie
de
conduction
accessoire
atrioventriculaire.
Les
caractéristiques
de
ce
type
de
tachycardie
sont
résu-
mées
dans
le
Tableau
1
(Fig.
1).
Si
cette
voie
accessoire
conduit
en
antérograde
en
rythme
sinusal,
l’espace
PR
est
court
et
le
QRS
«
empâté
»,
ce
qui
caractérise
un
syndrome
de
Wolff—Parkinson—White.
Quand
la
voie
de
conduction
accessoire
ne
conduit
qu’en
rétrograde,
l’ECG
en
rythme
sinusal
est
normal,
on
parle
alors
de
Kent
caché.
Ces
rythmes
réciproques
orthodromiques
sont
la
plus
fré-
quente
des
TSV
des
premières
années
de
vie,
décrite
dans
85
%
des
cas
avant
1
an.
L’évolution
de
ces
formes
précoces
est
favorable.
Passé
l’âge
de
12
à
18
mois,
les
crises
cessent.
Jusqu’à
93
%
des
enfants
ne
font,
en
effet,
pas
de
nouvelle
crise
après
1
an.
Cependant
le
suivi
à
moyen
et
long
terme
montre
des
récurrences
de
la
tachycardie
allant
de
30
à
60
%
selon
la
durée
du
suivi
[3,4].
La
prise
en
charge
initiale
consiste,
si
nécessaire,
en
le
rétablissement
des
fonctions
vitales
en
réanimation
et
la
réduction
du
trouble
du
rythme.
Passé
cette
période,
le
risque
résiduel
est
celui
de
la
récidive
qui,
chez
le
nouveau-né
et
nourrisson,
peut
conduire
à
une
cardiomyo-
pathie
dilatée
hypokinétique
si
la
tachycardie
n’est
pas
reconnue
et
réduite
à
temps.
Il
est
donc
coutume
de
pré-
venir
les
récidives
pendant
les
12
premiers
mois
de
vie.
Le
choix
de
la
molécule
relève
généralement
d’habitudes
personnelles.
Les
antiarythmiques
les
plus
couramment
utilisés
sont
les
bêtabloquants,
la
flécaïnide,
la
digoxine
(sauf
en
cas
de
syndrome
de
Wolff—Parkinson—White
avéré)
et
l’amiodarone
dont
les
modalités
de
prescription
sont
résumées
dans
le
Tableau
2.
L’utilisation
des
inhibiteurs
calciques,
notamment
par
voie
intraveineuse,
est
contre-
indiquée
chez
l’enfant
de
moins
de
2
ans.
Ces
traitements
peuvent
être
interrompus
après
le
premier
anniversaire,
qu’il
y
ait
ou
non
un
syndrome
de
Wolff—Parkinson—White
sur
l’ECG.
À
l’arrêt
du
traitement,
les
parents
doivent
être
formés
à
reconnaître
les
symptômes
de
tachycar-
die
et
éventuellement
savoir
prendre
le
pouls
de
leur
enfant.
En
cas
de
récidive
ou
de
première
crise
au-delà
de
la
première
année,
les
options
thérapeutiques
sont
plus
larges
et
dépendent
de
la
fréquence
et
de
la
tolé-
rance
des
accès
de
tachycardie.
L’ablation
par
cathéter
est
habituellement
proposée
aux
«
grands
enfants
».
Il
existe
cependant
quelques
rares
indications
d’ablation
avant
5
ans,
voire
dans
les
premiers
mois
de
vie
en
cas
de
tachycardies
réfractaires
compromettant
le
pronostic
vital
de
l’enfant
[5].
Syndrome
de
Wolff—Parkinson—White
Le
syndrome
de
Wolff—Parkinson—White
est
secondaire
à
la
présence
d’une
voie
de
conduction
accessoire
entre

Trouble
du
rythme
et
de
la
conduction
chez
l’enfant
193
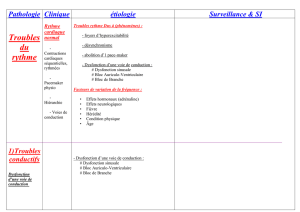
Tableau
1Caractéristiques
des
différentes
formes
de
tachycardies
supraventriculaires
(TSV).
Mécanisme
ECG
Rapport
P/QRS
Effet
des
manœuvres
vagales
Contexte
Épidémiologie
Population
Traitement
Figures
Tachycardies
jonctionnelles
RR
Réentrée
entre
le
nœud
auriculo-
ventriculaire
et
une
VA
patente
(syndrome
de
Wolff—Parkinson
—White)
ou
de
conduction
rétrograde
exclusive
(Kent
caché)
Tachycardie
régulière
à
QRS
fins.
FC
entre
260
et
300
bpm
chez
l’enfant
de
moins
de
1
an
et
160
à
250
chez
l’enfant
de
plus
de
1
an
P
=
QRS
Intervalle
R-
P>
65—70
ms
Réduction
de
la
tachycardie
Cœur
sain
le
plus
souvent
85
%
des
TSV
avant
1
an
Traitement
préventif
des
récidives
jusqu’à
1
an,
à
moyen
terme
RF
si
TJ
persiste
Fig.
1
PJRT Réentrée
entre
le
NAV
et
une
VA
de
conduction
rétrograde
décrémen-
tielle
le
plus
souvent
postéroseptale
droite
Tachycardie
régulière
entre
200
et
300
bpm
avant
2
ans
et
120—150
bpm
au-delà,
arrêt
et
redémarrage
spontané
incessant
P=
QRS
Pnégative
en
DII,
DIII
et
aVF
Aspect
de
R-P
long
Réduction
transitoire
de
la
tachycardie
Cœur
sain
Risque
de
car-
diomyopathie
dilatée
rythmique
<
1
%
des
TSV
de
l’enfant
Contrôle
difficile
de
la
tachycardie
nécessitant
souvent
l’association
de
plusieurs
antiaryth-
miques
Ablation
possible
Fig.
3
RIN
Réentrée
entre
deux
portions
du
NAV
de
vitesse
de
conduction
et
de
période
réfractaire
différentes
Tachycardie
régulière
à
QRS
fins
FC
entre
120
et
300
(typiquement
entre
180
et
250
bpm)
P
=
QRS,
P
rétrograde
juste
après
le
QRS
(<
65—70
ms),
peut
être
invisible
car
masquée
dans
le
complexe
ventriculaire
Réduction
de
la
tachycardie
Cœur
sain
Rare
avant
2
ans
Incidence
croissante
avec
l’âge
Traitement
antiaryth-
mique
jusqu’à
l’ablation
Fig.
4

194
A.
Maltret
Tableau
1
(
Suite
)
Mécanisme
ECG
Rapport
P/QRS
Effet
des
manœuvres
vagales
Contexte
Épidémiologie
Population
Traitement
Figures
Tachycardies
atriales
Flutter
Macroréentrée
intra-atriale,
le
plus
souvent
antihoraire
et
passant
par
l’isthme
cavotricuspide
(flutter
commun)
Tachycardie
régulière
de
220
à
300
bpm
Activité
atriale
en
«
dents
de
scie
»
bien
visible
en
inférieur
et
V1
(onde
F),
moins
caracté-
ristique
de
cas
de
flutter
cicatriciel
Fréquence
atriale
entre
300
et
500
chez
le
nouveau-né,
fréquence
ventriculaire
variable
selon
la
conduction
AV
en
1/1,
2/1,
3/1,
etc.
Fréquence
atriale
plus
lente
en
cas
de
flutter
cicatriciel,
conduction
en
1/1
plus
fréquente
Dégradation
de
la
conduction
AV
avec
démasquage
des
ondes
F
de
flutter
Nouveau-né
:
cœur
sain,
rare
Ebstein
Enfant
plus
grand
:
flutter
postatriotomie
et
dilatation
oreillette
droite
15
%
des
TSV
néonatales
Prédominance
masculine
Dépistage
et
réduction
anténatale
possibles
Nouveau-né
:
réduction
médicamenteuse
ou
électrique
Grand
enfant
:
ablation
par
radiofréquence
Fig.
5
Tachycardie
atriale
ectopique
Foyer
ectopique
de
l’oreillette
droite
ou
gauche
Régulier
après
possible
démarrage
et
fin
progressive
(warm-up
et
cool
down)
QRS
fins
P
>
QRS
ou
P
=
QRS
si
conduction
en
1/1
Dégradation
de
la
conduction
AV
(P
>
QRS)
sans
changer
fréquence
atriale
sous-jacente
Quelques
cas
de
réduction
de
la
tachycardie
avec
redémarrage
progressif
(warm-up)
Cœur
sain
Possible
car-
diomyopathie
dilatée
rythmique
2
%
des
TSV
néonatales
Traitement
antiarythmique
Ablation
possible
si
la
tachycardie
atriale
persiste
avec
l’âge
Fig.
6
Tachycardie
atriale
hissienne
Foyer
ectopique
du
faisceau
de
His
Tachycardie
régulière
à
QRS
fins,
le
plus
sou-
vent
<
200
bpm
P
<
QRS
avec
dissociation
AV
ou
P
=
QRS
si
conduction
rétrograde
en
1/1
Ne
change
pas
la
cadence
ventriculaire
Permet
d’objectiver
la
dissociation
AV
si
conduction
rétrograde
1/1
préalable
Cœur
sain,
forme
familiale
avec
risque
de
car-
diomyopathie
dilatée
rythmique
ou
postopératoire
Rare
Contrôle
difficile
de
la
tachycardie
par
les
antiarythmiques,
dont
l’objectif
est
de
ralentir
le
foyer
Ablation
difficile
Fig.
7
ECG
:
électrocardiogramme
;
VA
:
voie
accessoire
;
FC
:
fréquence
cardiaque
;
bpm
:
battements/min
;
TSV
:
tachycardies
supraventriculaires
;
TJ
:
tachycardies
jonctionnelles
;
RF
:
radiofréquence
;
RR
:
rythme
réciproque
;
RIN
:
réentrée
intranodale
;
PJRT
:
persistent
junctional
reciprocating
tachycardia
;
NAV
:
nœud
auriculo-ventriculaire
;
AV
:
auriculo-ventriculaire.

Trouble
du
rythme
et
de
la
conduction
chez
l’enfant
195
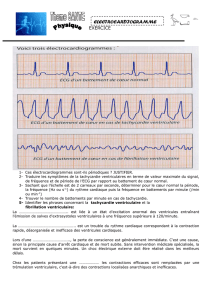
Figure
1.
A.
Tachycardie
jonctionnelle
réciproque
ou
«
rythme
réciproque
».
L’activité
atriale
rétrograde
est
bien
visible
dans
les
précordiales.
B.
Arrêt
brusque
de
la
tachycardie.
l’oreillette
et
le
ventricule.
En
rythme
sinusal,
le
passage
de
l’influx
électrique
par
la
voie
nodohissienne
et
par
la
voie
de
conduction
accessoire
produit
l’aspect
empâté
du
QRS
encore
appelé
onde
delta.
Hormis
la
survenue
d’accès
de
réentrée
décrit
ci-dessus,
le
risque
du
syndrome
de
Wolff—Parkinson—White
est
celui
d’une
mort
subite
consécutive
à
la
conduction
rapide
d’une
fibrillation
atriale
aux
ventricules
(Fig.
2),
dégéné-
rant
en
fibrillation
ventriculaire.
Ce
risque
est
dépendant
des
propriétés
électrophysiologiques
spécifiques
à
chaque
voie
de
conduction
accessoire.
Le
risque
de
fibrillation
atriale
est
souvent
lié
à
l’effort
chez
l’enfant
mais
son
incidence
est
considérée
comme
très
faible
avant
l’âge
de
10
ans.
L’exploration
de
la
perméabilité
antérograde
de
la
voie
de
conduction
accessoire
commence
par
une
épreuve
d’effort
et
un
Holter
qui,
s’ils
montrent
un
affinement
du
QRS,
permettent
de
conclure
à
la
bénignité
de
la
voie
de
conduction
accessoire.
Pourtant,
dans
la
majorité
des
cas
(85),
ces
examens
ne
permettent
pas
de
conclure
formelle-
ment.
On
peut
alors
proposer
une
exploration
œsophagienne
ou,
comme
c’est
le
cas
de
plus
en
plus
souvent,
une
explo-
ration
endocavitaire
qui
sera
complétée
par
une
ablation
de
la
voie
de
conduction
accessoire
si
celle-ci
est
maligne
et/ou
symptomatique
et
à
distance
des
voies
de
conduc-
tion
normales.
On
retient
donc
que
tout
syndrome
de
Wolff—Parkinson—White,
même
asymptomatique,
doit
être
exploré
avant
l’entrée
au
collège.
Tachycardie
jonctionnelle
réciproque
permanente
ou
tachycardie
de
Coumel
La
tachycardie
jonctionnelle
réciproque
permanente
ou
per-
sistent
junctional
reciprocating
tachycardia
(PJRT)
est
une
forme
rare
mais
potentiellement
grave
de
TSV
de
l’enfant.
Il
s’agit
d’une
réentrée
entre
le
nœud
auriculo-ventriculaire
et
une
voie
accessoire
spécifique
dans
ses
propriétés
électro-
physiologiques
et
sa
localisation.
La
conduction
dans
cette
voie
de
conduction
accessoire
est
lente
donc
facilement
per-
méable.
La
tachycardie
est
quasi-permanente,
entrecoupée
de
passage
spontané
en
rythme
sinusal
plus
ou
moins
long.
L’aspect
ECG
est
typique
comme
résumé
dans
le
Tableau
1
(Fig.
3).
Le
caractère
permanent
de
la
tachycardie
expose
au
risque
de
cardiomyopathie
rythmique.
De
petites
séries
de
la
littérature
rapportent
jusqu’à
50
%
de
dysfonc-
tion
ventriculaire
gauche
chez
les
enfants
avec
PJRT
[6].
C’est
une
tachycardie
difficile
à
contrôler
par
les
anti-
arythmiques
;
l’amiodarone
est
le
traitement
de
première
intention
[7].
Une
bi-,
voire
une
trithérapie
peut
être
néces-
saire.
L’ablation
par
radiofréquence
est
possible
et
efficace.
Tachycardie
jonctionnelle
par
réentrée
intranodale
Elle
est
considérée
comme
rare
dans
la
première
année
de
vie.
Pourtant,
selon
les
séries,
elle
est
rapportée
dans
3
à
19
%
des
tachycardies
jonctionnelles
avant
1
an
[8].
C’est
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
1
/
20
100%