SOMMAIRE

A
AS
SS
SO
OC
CA
AT
TI
IO
ON
N
D
DE
ES
S
P
PR
RA
AT
TI
IC
CI
IE
EN
NS
S
D
DE
E
G
GE
EN
NE
ET
TI
IQ
QU
UE
E
M
MO
OL
LE
EC
CU
UL
LA
AI
IR
RE
E
(
(A
AN
NP
PG
GM
M)
)
SCLEROSE TUBEREUSE DE BOURNEVILLE
Page : 1/14
Référence : ANPGM_ 018 Numéro de version : 1.0
Date de Création : 01/03/2009
Date de validation en assemblée plénière: 15/06/2009
Date de la remise à jour :
Motif :
Validation :
Nom
Hôpital
Date
Rédacteur(s)
Marie claire Malinge
Renaud Touraine
Laboratoire de Génétique
Moléculaire -CHU - ANGERS
Laboratoire de Génétique
Moléculaire -CHU – Saint Etienne
01/03/2009
Vérificateur(s)
Sous-groupe Neuromusculaire
01/03/2009
Approbateur(s)
Bureau ANPGM :
Michel GOOSSENS
Marc DEPLECH
Michel VIDAUD
Hôpital Henri Mondor-APHP
Hôpital Cochin-APHP
Hôpital Beaujon-APHP
15/06/2009

A
AS
SS
SO
OC
CA
AT
TI
IO
ON
N
D
DE
ES
S
P
PR
RA
AT
TI
IC
CI
IE
EN
NS
S
D
DE
E
G
GE
EN
NE
ET
TI
IQ
QU
UE
E
M
MO
OL
LE
EC
CU
UL
LA
AI
IR
RE
E
(
(A
AN
NP
PG
GM
M)
)
SCLEROSE TUBEREUSE DE BOURNEVILLE
Page : 2/14
Référence : ANPGM_ 018 Numéro de version : 1.0
SOMMAIRE
I. Rappels sur la pathologie.......................................................................................................... 3
II. Prérequis cliniques pour l'analyse génétique ......................................................................... 4
A. Proposant ............................................................................................................................................ 3
B. Conseil génétique des apparentés d'un sujet atteint .......................................................................... 3
C. Diagnostic prénatal ............................................................................................................................. 4
III. Arbres décisionnels pour l'analyse génétique ....................................................................... 6
A. Analyses génétiques ........................................................................................................................... 6
B. Arbres décisionnels pour un diagnostic postnatal .............................................................................. 8
C. Arbres décisionnels pour le conseil génétique ................................................................................. 10
Annexe : Liste des laboratoires effectuant le diagnostic moléculaire de la sclérose
tubéreuse de Bourneville ........................................................................................................... 12

A
AS
SS
SO
OC
CA
AT
TI
IO
ON
N
D
DE
ES
S
P
PR
RA
AT
TI
IC
CI
IE
EN
NS
S
D
DE
E
G
GE
EN
NE
ET
TI
IQ
QU
UE
E
M
MO
OL
LE
EC
CU
UL
LA
AI
IR
RE
E
(
(A
AN
NP
PG
GM
M)
)
SCLEROSE TUBEREUSE DE BOURNEVILLE
Page : 3/14
Référence : ANPGM_ 018 Numéro de version : 1.0
I. Rappels sur la pathologie
La sclérose tubéreuse de Bourneville (STB) est une maladie génétique, caractérisée par le développement de tumeurs
bénignes, qui peuvent toucher différents organes: la peau, le cerveau, les reins, les yeux, le coeur, et les poumons. Cette maladie
présente une grande diversité de manifestations avec une évolution très variable d’une personne à l’autre, allant de formes
pratiquement indétectables, à des formes plus sévères. Elle a été décrite pour la première fois en 1880 par D.M. Bourneville.
La prévalence de la STB dans la population générale est de 1 sur 7 000 à 1 sur 10 000. La STB, maladie autosomique dominante, peut
toucher aussi bien les filles que les garçons, quelle que soit leur origine géographique. Pour la STB, dans 2/3 des cas, il s'agit d'une
forme sporadique. Le diagnostic peut être posé avant la naissance si lors de l'échographie prénatale des tumeurs cardiaques sont
détectées chez le fœtus. Mais le diagnostic peut être porté plus tard, chez le nourrisson, dans l’enfance ou à l’âge adulte.
Les manifestations de la STB sont très variées:
1. Manifestations cutanées:
- Les angiofibromes faciaux ou adénomes de Pringle présents chez 85 % des personnes atteintes. Ils apparaissent surtout après l’âge
de deux ans.
- Les taches achromiques ou hypomélaniques retrouvées chez 80 % des personnes atteintes. Elles apparaissent souvent précocement,
dès la première année de vie. Cependant, elles ne sont pas spécifiques et peuvent se voir chez des personnes n’ayant pas de STB.
- La « plaque fibreuse du front » présente chez 25 % des personnes atteintes. Elle peut apparaître tôt dans la vie.
- Les plaques « peau de chagrin » présentes chez 20 à 40 % des malades. Elles peuvent être présentes à la naissance mais
surviennent souvent plus tard.
- Les fibro-kératomes unguéaux (tumeurs de Koenen) se voient chez 50 % des malades. Ils apparaissent assez tardivement et sont
donc rarement présents chez l’enfant.
- Les molluscum pendulum retrouvés chez 30 % des personnes atteintes.
2. Atteintes du système nerveux:
Elles sont variables et peuvent se manifester par une épilepsie, une déficience intellectuelle ou des troubles du comportement.
- L’épilepsie est fréquente et se rencontre chez 60 à 80 % des personnes atteintes. Les crises peuvent survenir très tôt dans la vie,
chez le nourrisson, où elles se manifestent souvent sous la forme de spasmes appelés syndrome de West qui n’est cependant pas
spécifique de la STB. Les crises d’épilepsie peuvent aussi commencer plus tard dans la vie. L’épilepsie est essentiellement liée à la
présence dans le cerveau des personnes atteintes de tubers corticaux. Toutefois, il est possible d’avoir des tubers sans faire de crise
d’épilepsie. Il existe aussi souvent d’autres tumeurs cérébrales appelées nodules sous-épendymaires.
- Bien que plus de la moitié des personnes présentant une STB n’aient pas de déficience intellectuelle, certains enfants rencontrent des
difficultés d’apprentissage. Dans certains cas, un déficit intellectuel plus important peut même être présent surtout chez les enfants qui
ont eu une épilepsie précoce, en particulier un syndrome de West.
Les troubles du comportement sont fréquents et variés : de type autistique, hyperactivité avec déficit de l’attention, comportement
agressif, troubles du sommeil… et leur intensité diffère d’une personne à l’autre.
3. Atteinte de l’œil:
Des tumeurs bénignes de la rétine appelés phacomes rétiniens, sont présentes chez environ la moitié des personnes ayant une STB.
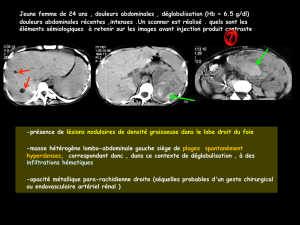
4. Atteinte des reins:
Cette atteinte est assez fréquente, surtout après l’âge de 10 ans et chez l’adulte. Elle est rare chez le petit enfant. Elle consiste surtout
en la présence de tumeurs rénales bénignes, (angiomyolipomes).
5. Atteinte du cœur:
Elle est constituée avant tout par la présence d’une ou plusieurs tumeurs cardiaques (rhabdomyomes). Elles peuvent être présentes
avant la naissance et peuvent être découvertes lors de l’échographie prénatale.
6. Atteinte des poumons:
Cette atteinte touche presque exclusivement la femme adulte et cette forme est appelée lymphangioléiomyomatose.
7. Autres atteintes:
D’autres organes peuvent être touchés comme le tube digestif, les os… Des atteintes sont aussi plus rares telles que: une hypertrophie
au niveau des gencives et de très fins sillons dans l’émail dentaire
8. L’évolution de la maladie est extrêmement variable d’une personne à l’autre, y compris au sein d’une même famille où les
malades sont pourtant porteurs d’une anomalie génétique identique. Certaines complications tumorales rénales ou cérébrales, des
crises d’épilepsie prolongées (état de mal épileptique) ou bien une atteinte pulmonaire peuvent conduire à un décès prématuré.
Pour la STB, deux gènes ont été identifiés. Le premier est le gène TSC1, localisé sur le chromosome 9. Il code pour
l’hamartine. Le second gène TSC2, est situé sur le chromosome 16 et code pour la tubérine. Ces deux gènes sont des gènes
suppresseurs de tumeurs. L’hamartine et la tubérine doivent s’associer pour être actives ; si l’une des deux protéines est absente ou
anormale, le complexe hamartine-tubérine ne se forme pas ou est inactif. Il n’y a pas de différence notable au niveau des
manifestations clinique entre les personnes ayant une maladie liée à une mutation de TSC1 et celle ayant une forme liée à TSC2. Les
mutations des gènes TSC1 et TSC2 sont très nombreuses et variées et sont difficiles à mettre en évidence.
Modifié d'après la source: Orphanet, auteurs: Marie-Claire Malinge, Docteur Renaud Touraine, Professeur Jean-François Cordier,
Professeur Pierre Wolkenstein, Association Française des Conseillers en Génétique, Association Sclérose Tubéreuse de
Bourneville; www.orpha.net/data/patho/pub/fr/sclerosetubereusebourneville-FRfrPub660v01.pdf Mai 2007

A
AS
SS
SO
OC
CA
AT
TI
IO
ON
N
D
DE
ES
S
P
PR
RA
AT
TI
IC
CI
IE
EN
NS
S
D
DE
E
G
GE
EN
NE
ET
TI
IQ
QU
UE
E
M
MO
OL
LE
EC
CU
UL
LA
AI
IR
RE
E
(
(A
AN
NP
PG
GM
M)
)
SCLEROSE TUBEREUSE DE BOURNEVILLE
Page : 4/14
Référence : ANPGM_ 018 Numéro de version : 1.0
II. Contexte clinique pour l'analyse génétique
A. Proposant
Le diagnostic de la STB se fonde avant tout sur les manifestations cliniques. Le plus souvent, ce diagnostic
n’est pas très difficile à poser, car à l’âge adulte, 95 % des personnes présentent des signes évocateurs. Il
peut, cependant, être moins facile chez l’enfant, ayant des manifestations plus discrètes.
Les examens complémentaires vont aider au diagnostic lorsque les manifestations cliniques sont discrètes:
- L’examen de la peau sous lumière de Wood permettra de mettre en évidence les taches
achromiques.
- Le fond d’oeil recherchera des phacomes rétiniens.
- L’échographie rénale permettra de mettre en évidence des tumeurs et des kystes et l’échographie
cardiaque des rhabdomyomes.
- Un examen en résonance magnétique nucléaire (IRM) peut aussi être proposé. Cet examen permet
de rechercher la présence de lésions caractéristiques de la STB (tubers corticaux ou nodules sous-
épendymaires).
De nombreuses manifestations présentes dans la STB ne sont pas spécifiques de la maladie et peuvent être
tout à fait isolées ou être le symptôme d’une autre affection. C’est le cas notamment des taches
dépigmentées qui peuvent s’observer chez des personnes indemnes de toute maladie ou entrer dans le
cadre de maladies de la peau (vitiligo, piébaldisme…).
Les tumeurs rénales et cardiaques et l’atteinte pulmonaire peuvent aussi survenir chez des personnes
n’ayant pas de STB. Par conséquent, c’est surtout l’association de plusieurs manifestations différentes plus
que ces manifestations prises isolément, qui permet d’établir le diagnostic de la maladie.
La maladie peut être dépistée dès la naissance ou plus tardivement. Pour ce dépistage, l’examen clinique
approfondi et les examens complémentaires décrits plus haut sont nécessaires. Si l’anomalie génétique a
été mise en évidence, il est possible de la rechercher chez les apparentés de la personne atteinte.
B. Conseil génétique des apparentés d'un sujet atteint
La STB est une affection autosomique dominante, ce qui signifie qu’elle peut se transmettre de génération
en génération. A chaque grossesse, une personne atteinte a un risque sur deux de transmettre la maladie à
ses enfants, quel que soit leur sexe. Une personne non atteinte ne transmettra pas la maladie.
Dans deux cas sur trois, l’enfant atteint est né de deux parents indemnes. Il s’agit alors d’une néomutation.
Cependant, on ne peut affirmer qu’il s’agit d’une néomutation que si les deux parents ont subi d’une part, un
examen clinique soigneux avec des examens complémentaires et d’autre part, une analyse moléculaire du
gène, afin d’éliminer chez eux une forme légère sans symptômes apparents. Si aucun des deux parents
n’est porteur de la mutation, le risque théorique d’avoir un deuxième enfant atteint est de l’ordre de 1 à 2 %
en raison d’un phénomène rare: la mosaïque germinale. La maladie peut alors réapparaître chez un
deuxième enfant, alors que les parents ne sont pas atteints par la maladie. Il n’est pas possible de mettre en
évidence cette mosaïque germinale et par conséquent, de distinguer les couples qui n’ont aucun risque de
transmettre la maladie. Dans ce cas, un diagnostic prénatal peut être proposé au couple même si le risque
est faible.
Par ailleurs, la STB est une maladie d’expressivité très variable ce qui signifie que les manifestations
cliniques sont différentes d’une personne à l’autre notamment sur le plan de la gravité. Une personne
porteuse du gène muté a forcément des signes, mêmes minimes (par exemple, des lésions au fond d’oeil,
au cerveau ou aux reins).

A
AS
SS
SO
OC
CA
AT
TI
IO
ON
N
D
DE
ES
S
P
PR
RA
AT
TI
IC
CI
IE
EN
NS
S
D
DE
E
G
GE
EN
NE
ET
TI
IQ
QU
UE
E
M
MO
OL
LE
EC
CU
UL
LA
AI
IR
RE
E
(
(A
AN
NP
PG
GM
M)
)
SCLEROSE TUBEREUSE DE BOURNEVILLE
Page : 5/14
Référence : ANPGM_ 018 Numéro de version : 1.0
Les frères et soeurs d’un malade ont un risque sur deux d’être également atteints si l’un des deux parents a
la maladie. En revanche, si les parents sont indemnes, et qu'il s'agit d'une néomutation chez l’enfant atteint,
le risque pour les frères et soeurs peut être considéré très faible (très rares cas de mosaïque germinale). Les
frères et sœurs indemnes ne transmettent pas l’affection à leur descendance.
C. Diagnostic prénatal
Le diagnostic prénatal peut être réalisé dans certaines conditions. Cependant, le diagnostic prénatal dans
cette affection pose un problème car la gravité de la maladie est extrêmement variable, y compris au sein
d’une même famille. Il est impossible de prédire à l’avance la sévérité de la maladie. Chaque cas est
particulier et les familles concernées doivent être vues en consultation de conseil génétique afin d'évaluer le
risque pour un couple d'avoir un enfant atteint.
Le prélèvement des villosités choriales a l’avantage de se pratiquer plus tôt au cours de la grossesse. Ces
examens entraînent un risque faible de fausse couche, différent selon le choix de la technique de
prélèvement, qu’il convient de discuter en consultation de génétique au préalable
Plusieurs situations sont possibles :
- Dans la première situation, il y a au moins une personne atteinte de STB dans la famille.
Deux cas peuvent alors se présenter :
. Si la mutation est connue dans la famille et que le parent à risque a été testé, le diagnostic prénatal
est réalisable par analyse moléculaire du gène, tout en sachant que les manifestations de la maladie varient
d’une personne à l’autre. Les deux techniques de prélèvement utilisées sont l’amniocentèse et le
prélèvement des villosités choriales.
. Si la mutation n’est pas connue, le diagnostic prénatal est éventuellement possible, mais
tardivement par la surveillance échographiques pour les tumeurs cardiaques et la réalisation d’une IRM
cérébrale foetale à 30 semaines d’aménorrhée. Cependant, ces examens du coeur et du cerveau ne
permettent pas de déceler tous les cas de STB avant la naissance.
- Dans la deuxième situation, il n’y a pas de cas dans la famille et l’échographie prénatale a mis en
évidence chez le foetus des rhabdomyomes cardiaques, permettant d’évoquer le diagnostic de STB. Il faut
alors réaliser une IRM cérébrale et une étude moléculaire des gènes TSC1 et TSC2 peut être proposée si
une amniocentèse est réalisée.
 6
7
8
9
10
11
12
13
14
6
7
8
9
10
11
12
13
14
1
/
14
100%