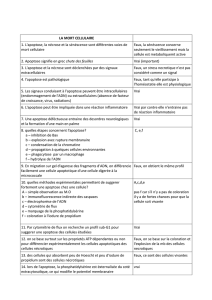
L`apoptose ou mort cellulaire programmée est une forme

L'apoptose ou mort cellulaire programmée est une forme physiologique de mort
cellulaire hautement régulée et elle est nécessaire à la survie des organismes
multicellulaires. Typiquement, les cellules d'organismes multicellulaires
s'autodétruisent lorsque celles-ci ne sont plus utiles, lorsqu'elles sont
endommagées ou lorsqu'elles sont dysfonctionnelles. L'apoptose implique
habituellement des cellules individuelles dans un tissu et ne provoque pas
l'inflammation.
Il existe principalement 2 voies d'induction de l'apoptose dans la cellule: la voie
mitochondriale est induite par la relâche de protéines mitochondriales et la voie
des récepteurs à domaine de mort situé dans la membrane plasmique.
Lors de l'apoptose, les cellules sont d'abord modifiées au niveau physiologique. Le
premier point de non retour dans l'apoptose induite par la voie mitochondriale est la
relâche de cytochrome c de la mitochondrie et la dégradation du potentiel
membranaire mitochondrial. Les caspases sont ensuite activées et celles-ci
provoquent la coupure des protéines associées à l'apoptose. Les
phosphatidylsérines sont ensuite transloquées de la face interne de la membrane
cellulaire à la face externe. Les caractéristiques morphologiques de l'apoptose
comme la condensation du cytoplasme, du noyau et de la chromatine, une
fragmentation de l'ADN, un bourgeonnement de la membrane plasmique ainsi
qu'une perte de l'asymétrie membranaire sont alors visibles. Il y a ensuite formation
de corps apoptotiques qui sont digérés par les macrophages environnants.
Schéma simplifié représentant les protéines essentielles au programme de mort cellulaire chez
C.elegans et chez les mammifères.

I - MÉCANISMES MOLÉCULAIRES DE L'APOPTOSE : ASPECT GÉNÉRAL
I-3. Les Caspases
a) Nomenclature
Comme nous l'avons déjà noté, ced-3 code pour une protéase à cystéine homologue à ICE (Thornberry et al.,
1992). Les protéases apoptogènes sont des protéases à cystéine qui possèdent une spécificité stricte de clivage de
leur substrats après un résidu d'acide aspartique. Cette spécificité de clivage n'est partagée qu'avec une seule
autre protéase, le granzyme B (une sérine protéase présente dans les lymphocytes T cytotoxiques).
Une nouvelle nomenclature proposée par Alnemri et coll. regroupe désormais les protéases apoptogènes sous le
nom de CASPASE (TABLE 1). Le C représente la cystéine du centre actif (QACxG) et aspase définit la
spécificité stricte de clivage des substrats de cette famille de protéases après un acide aspartique. L'ICE, qui fut
chronologiquement la première caspase caractérisée, a donc été tout naturellement rebaptisée caspase 1. À ce
jour 14 caspases ont été identifiées mais il ne fait aucun doute que cette liste n'est pas exhaustive.
b) Structure
Toutes les caspases ont une structure très conservée comprenant, un prodomaine N-terminal de taille variable, un
domaine qui deviendra après clivage la grande sous-unité (17-21 kDa, qui porte le centre actif) et un domaine qui
deviendra après clivage la petite sous-unité (10-14 kDa). Certains membres de la famille des caspases possèdent
un domaine de liaison entre la grande et la petite sous-unité. Les prodomaines sont variables, à la fois dans leur
taille et dans leur séquence. Ainsi les caspases 3, 6 et 7 ont un petit prodomaine alors que les caspases 1, 2, 4, 5,
8, 9, 10, 11, 12 et 13 possèdent un grand prodomaine. Les caspases à petits prodomaines sont souvent regroupées
sous le nom de caspases effectrices. Ces caspases sont activées par des caspases dites initiatrices. Les
prodomaines semblent jouer un rôle dans les interactions protéines-protéines. Ainsi les prodomaines des
caspases 8 et 10 contiennent des Domaines Effecteurs de Mort Cellulaire (ou Death Effector Domains : DEDs)
qui sont des structures permettant la liaison de la caspase aux molécules adaptatrices FADD (Boldin et al., 1995;
Chinnaiyan et al., 1995) ou TRADD (Hsu et al., 1995) (cf. chapitre I-5). Certaines autres caspases (caspases 1, 2,
4 et 9) possèdent un Domaine de Recrutement des Caspases (ou Caspase Recruitment Domain : CARD
(Hofmann et al., 1997)). Ces CARDs jouent un rôle dans l'interaction entre caspases ainsi qu'avec une grande
variété de molécules adaptatrices ou régulatrices (cf. chapitre I-5)
c) Activation
Figure 2
agrandissement
Structure et activation des caspases.
La conversion de la caspase à l'état de zymogène en une enzyme mature nécessite au moins deux clivages au
niveau de liaison Asp-X (TABLE 1). Ces clivages successifs ont lieu de manière séquencielle : tout d'abord
coupure entre la grande et la petite sous-unité (donc il y a libération de la petite sous-unité du reste de la
molécule) suivie par la libération du prodomaine (Figure 2). La caspase va alors pouvoir s'assembler sous sa
forme active, composée de deux grandes et de deux petites sous-unités.
La structure générale ainsi obtenue est (p10/p20)2 (Walker et al., 1994; Wilson et al., 1994; Rotonda et al.,
1996). Les caspases vont pouvoir s'auto-activer et/ou être activées par d'autre caspases. Cette remarque introduit
la notion de cascade d'activation. Ainsi une fois les caspases initiatrices activées, elles vont pouvoir cliver
d'autres caspases encore à l'état de zymogène (notamment les caspases effectrices). Ce type d'activation en
cascade permet probablement la régulation et l'amplification du signal.
Cette suite d'événements est généralement divisée en trois étapes :
- une étape d'induction qui est réversible

- une étape d'exécution qui est régulable
- une étape de dégradation qui est irréversible.
Certaines études ont décrit la possibilité qu'une sous-unité p10 issue d'un zymogène puisse s'associer avec la
sous-unité p20 d'un autre zymogène durant la maturation des précurseurs (Walker et al., 1994; Wilson et al.,
1994; Rotonda et al., 1996). Il est à noter que certaines protéases n'ayant pas de spécificité pour un acide
aspartique sont capables d'activer les zymogènes des caspases au moins in vitro (Zhou et Salvesen, 1997). Ce
clivage intervient au niveau de sites alternatifs situés dans le segment de liaison entre la grande et la petite sous-
unité. De plus il a été décrit que la caspase 9 pouvait être activée sans clivage (Stennicke et al., 1999).
L'activation et l'activité des caspases peuvent être modulées de différentes façons (par phosphorylation, par
exemple). Ce point sera détaillé dans le chapitre I-3-f.
d) Substrats
- GÉNÉRALITÉS
Figure 3
agrandissement
Structure tridimentionnelle des caspases.
Le rôle des caspases est principalement exécutif, c'est-à-dire qu'elles vont s'attacher à éteindre les voies
protectrices et à activer des molécules qui vont participer à la destruction cellulaire.
Les caspases sont des enzymes extrêmement sélectives. Ainsi, une analyse sur gel bi-dimentionnel d'extraits
obtenus à partir de cellules vivantes ou bien de cellules en apoptose n'a révélé que de faibles modifications dans
le profil global ( Robaye et al., 1994; Amess et Tolkovsky, 1995; Gerner et al., 1998; Kaufmann, 1989). Les
protéines cibles doivent impérativement posséder un aspartate en position P1 (Sleath et al., 1990; Howard et al.,
1991). Cet aspartate sera niché dans une poche (désignée site S1) et sera ainsi alignée avec Arg179, Gln283,
Arg341 et Ser347 (numérotation correspondant à la caspase 1). Cette structure est conservée chez toutes les
caspases humaines (à l'exception de Ser347 qui est remplacée par une Thr dans la caspase 8) (Figure 3).
La TABLE 2 indique les principaux substrats clivés par les caspases au cours de l'apoptose. Ces protéines cibles
regroupent des protéines cytoplasmiques, nucléaires, des protéines impliquées dans le métabolisme et la
réparation de l'ADN et des protéines kinases. De plus, des protéines impliquées dans la transduction du signal et
dans l'expression de gènes, dans la régulation du cycle cellulaire, la prolifération, dans les maladies génétiques
ou des protéines de régulation de l'apoptose sont aussi substrats des caspases (cf. TABLE 2).
Il est toutefois à noter que certains substrats ne sont pas clivés dans tous les types cellulaires. L'actine, par
exemple, est clivée dans la lignée myélomonocytaire U937 (Mashima et al., 1997), dans les neurones (Villa et
al., 1998) et dans les thymocytes (Villa et al., 1998) mais pas dans les autres types cellulaires durant l'exécution
du programme apoptotique (Song et al., 1997; Rice et al., 1998). De plus, certains substrats sont clivés à des sites
différents selon le type cellulaire. Ainsi, la topoisomérase I a un profil de clivage différent selon qu'il s'agisse de
cellules de cancer de poumon (A549) ou de cellules de cancer du sein (MDA-MB-468) (Samejima et al., 1999).
Cette hétérogénéité pourrait soit refléter l'activation de caspases différentes, soit des variations dans
l'accessibilité des substrats par les protéases, soit une combinaison de ces deux hypothèses.
Etant donné l'hétérogénéité et le nombre des substrats clivés par les caspases (TABLE 2), j'ai choisi, pour des
raisons de clarté et d'adéquation à mon travail de recherche, de ne détailler que les kinases substrats des caspases
au cours de l'apoptose.
- PROTÉINES KINASES
Au moins 13 protéines kinases sont clivées durant l'apoptose (cf. TABLE 2). La majeure partie de ces kinases le
sont dans leurs domaines régulateurs (ou juste à coté), produisant des formes tronquées constitutivement actives.

Ce type de régulation concerne les kinases suivantes : MEKK1 (Cardone et al., 1997; Deak et al., 1998;
Widmann et al., 1998), PAK2 (Lee et al., 1997; Rudel and Bokoch, 1997), Mst1/Krs et Mst2 (Graves et al.,
1998; Lee et al., 1998), PKCd (Ghayur et al., 1996), PKCq (Datta et al., 1997), PKCb1 (Shao et al., 1997) et
PRK2 (Cryns et al., 1997). L'expression des formes tronquées de ces kinases induit une apoptose massive des
cellules (Ghayur et al., 1996; Datta et al., 1997; Lee et al., 1997; Cardone et al., 1997; Graves et al., 1998;
Widmann et al., 1998). Par exemple, pour les kinases MEKK1, PAK2 et Mst1, l'expression du fragment activé
par clivage est pro-apoptotique alors que l'expression d'une forme inactive retarde l'apoptose (Lee et al., 1997;
Rudel et Bokoch, 1997 ; Cardone et al., 1997; Widmann et al., 1998). Ces résultats semblent indiquer que ces
trois kinases ont pour cibles des protéines essentielles pour la régulation de l'apoptose. Il convient de signaler
que ces kinases sont toutes capables d'activer la voie SAPK/JNK (Stress Activated Protein Kinase/Jun N-
terminal Kinase) qui conduit à l'exacerbation de la transcription de gènes placés sous le contrôle du facteur de
transcription c-Jun. Nous pouvons donc envisager que ces kinases sont clivées dans le but d'activer SAPK/JNK
(kinase impliquée dans l'apoptose, (Graves et al., 1998; Lee et al., 1998) (Cardone et al., 1997; Deak et al.,
1998). Ceci est d'autant plus vraisemblable que la voie antagoniste, (la voie ERK1 et 2 (Xia et al., 1995)) est
régulée négativement durant l'apoptose.
Certains membres de la famille des PKC sont également constitutivement activés par clivage au cours de
l'apoptose. Ainsi l'expression de fragments actifs des PKCd et PKCq induit l'apoptose des cellules transfectées
(Datta et al., 1997; Ghayur et al., 1996).
Durant ce travail de thèse nous avons identifié la tyrosine kinase p59Fyn comme nouveau substrat des caspases
(cf. partie résultat).
e) Invalidation génique
Etant donné le grand nombre de caspases ainsi que l'absence d'inhibiteur réellement sélectif d'une caspase
donnée, l'implication individuelle de ces protéases apoptogènes dans la mort cellulaire programmée n'a pu être
étudiée jusqu'à présent qu'en générant des animaux déficients pour l'expression de certaines d'entre elles. A ce
jour, seuls les gènes codant pour les caspases 1, 2, 3, 8, 9, 11 et 12 ont été invalidés (cf. TABLE 3).
- CASPASES 1 ET 11
Les souris caspase 1-/- et caspase 11-/- présentent un développement normal (Kuida et al., 1995; Li et al., 1995;
Wang S. et al., 1998). Cependant, elles présentent une production défectueuse d'IL-1a et b, d'IL-18 et
d'interferon-g. De plus, elles présentent une résistance accrue au choc septique. En fait, il semble bien établi que
la caspase 1 joue un rôle dans la régulation du système immunitaire mais pas ou peu dans les voies apoptotiques
(Kuida et al., 1995; Li et al., 1995). Récemment, il a été décrit que la caspase 1 est activée par une interaction
directe avec la caspase 11 (caspase murine) (Wang S. et al., 1998). La caspase 11 présente des homologies avec
les caspases humaines 4 et 5.
- CASPASE 2
Les souris caspase 2-/- présentent un développement normal jusqu'à l'âge adulte et ne présentent aucun
phénotype sévère (Bergeron et al., 1998). En revanche, la caspase 2 semble être requise pour la mort des cellules
germinales femelles. De plus, les ovocytes de ces souris présentent une résistance à l'apoptose induite par des
agents chimiothérapeutiques. A la naissance, les souris déficientes présentent une diminution du nombre de
motoneurones faciaux. Ceci nous indique que la caspase 2 n'agit pas simplement comme un effecteur positif de
l'apoptose mais qu'elle est aussi capable, selon le type cellulaire, de retarder la mort cellulaire. Cette différence
pourrait s'expliquer par la présence de deux formes de caspase 2 obtenues par épissage alternatif. Ainsi, il a été
montré que Casp2L (Long) induit l'apoptose alors que Casp2S (Small) inhibe l'apoptose (Wang L. et al., 1994).
Enfin, les lymphoblastes B de souris caspase 2-/- sont plus résistants à l'action combinée de la perforine et du
granzyme B mais ne présentent pas de sensibilité moindre à l'apoptose induite par un anticorps anti-Fas,
l'étoposide, la staurosporine ou les rayonnements g
En définitive, la caspase 2 est probablement essentielle pour l'apoptose des cellules germinales femelles mais elle
peut toutefois, dans certaines situations, avoir un effet protecteur contre l'apoptose. De plus, l'action de la caspase

2 semble être dépendante du type cellulaire, du stade de développement, de l'épissage de son ARNm ainsi que de
la présence ou de l'absence d'autres caspases.
- CASPASE 3
Les souris invalidées pour la caspase 3 furent les premières à présenter des profonds bouleversements de
l'apoptose (Kuida et al., 1996; Woo et al., 1998). Elles sont plus petites que les souris contrôles et meurent entre
la première et la troisième semaine après la naissance. De manière surprenante, il s'est avéré que le phénotype de
ces souris est extrêmement restreint. En effets, les anomalies les plus marqués semblent sélectivement localisés
au niveau du système nerveux central. Ainsi les animaux caspase 3-/- souffrent d'une hyperplasie cérébrale
massive. Les thymocytes issus de ces souris peuvent initier un programme apoptotique normal en réponse à un
anticorps anti-Fas, à la dexamethasone, au céramide-C2, à la staurosporine et aux rayonnement-g (Kuida et al.,
1996; Woo et al., 1998). En revanche, la caspase 3 semble être requise pour l'apoptose des neutrophiles et des
lymphocytes T activés (Woo et al., 1998). De plus, des études ont décrit que la caspase 3 n'était pas requise pour
le clivage de PARP (Kuida et al., 1996; Woo et al., 1998) ou la liaison à l'annexine V (qui reconnait
spécifiquement les résidus phosphatidylsérine) mais qu'elle était absolument nécessaire à la dégradation
internucléosomale de l'ADN ainsi qu'à la condensation de la chromatine (Woo et al., 1998). Il a été également
décrit que les lymphocytes T périphériques de ces souris étaient insensibles à l'AICD (Activation Induced Cell
Death, cf. chapitre III-2a) ainsi qu'à l'apoptose induite par un anticorps anti-CD3 (dirigé contre la partie
monomorphique du récepteur T) ou un anti-Fas (Woo et al., 1998).
En définitive, il semble que l'implication de la caspase 3 dépende du type de stimulus apoptotique considéré. De
plus, il existe une spécificité tissulaire. Ainsi, un traitement au TNFa induit une apoptose normale des
thymocytes issus de souris caspase 3-/- alors que les fibroblastes transformés y sont résistants (Woo et al., 1998).
Ceci souligne que la caspase 3 pourrait jouer un rôle différent selon le type de cellules et de stimuli considérés.
- CASPASE 8
Les souris caspase 8-/- se développent normalement durant les 11 premiers jours suivant la fécondation, puis
meurent, probablement des suites de malformations cardiaques importantes. Le cœur de ces animaux est
hypotrophique, suggèrant que la caspase 8 pourrait être impliquée dans la transmission des signaux de survie
plutôt que des signaux de mort, au moins au niveau de cet organe. De plus, ces embryons produisent très peu de
précurseurs myéloïdes (Varfolomeev et al., 1998). Les fibroblastes embryonnaires de ces souris sont insensibles
aux effets cytotoxiques initiés par Fas, TNF-RI ou DR3 mais restent sensibles à la déprivation en facteurs de
croissance, aux radiations U.V., au céramide et à l'étoposide (Varfolomeev et al., 1998). Les fibroblastes
embryonnaires répondent normalement aux signaux non apoptotiques émanant des récepteurs de mort et sont
capables d'activer la voie JNK ainsi que le facteur de transcription NF-kB de manière équivalente aux cellules
sauvages.
En définitive, ces résultats indiquent non seulement que la caspase 8 est un élément essentiel et non redondant de
l'apoptose initiée par les récepteurs de mort, mais aussi qu'elle joue un rôle essentiel (et à ce jour grandement
incompris) dans le développement cardiaque et dans l'hématopoïèse.
- CASPASE 9
Le phénotype de ces souris est semblable mais cependant plus sévère que celui des souris caspase 3-/- (V-
kuida98 et V-hakem98). Elles meurent au 16ème jour de développement. Elles souffrent de malformation
cérébrale avec un excès cellulaire au niveau du système nerveux central. On note environ 10 fois moins de
cellules TUNEL positives (i.e. apoptotiques) dans les cerveaux des souris invalidées au stade E12,5 par rapport à
des souris contrôles. Ce phénotype est aussi partagé par les souris Apaf-1-/- (cf. chapitre I-4). De plus, il existe
une absence d'activation de la (Hakem et al., 1998; Kuida et al., 1998) caspase 3 in vivo dans le cerveau mais
cette activation se produit normalement dans les tissus ectodermiques et méningés (Kuida et al., 1998).
Contrairement au cerveau, certains organes comme le cœur, le poumon, le foie mais aussi la colonne vertébrale
présentent un développement normal. Les cellules ES ou les fibroblastes embryonnaires de ces souris libèrent le
 6
7
8
9
10
11
12
13
6
7
8
9
10
11
12
13
1
/
13
100%