Protocole type d`essai thérapeutique

__________________________________
Equipe de Méthodes en recherche clinique
Sandrine Andrieu, Catherine Arnaud, Thierry Lang, Valérie Lauwers-Cancès
1
PROTOCOLE TYPE D’ETUDE DE COHORTE
1. Justification de l’étude ................................................................................................... 2
1.1. Définition de la maladie ou de l’événement étudié ................................................. 2
1.2. Connaissance scientifique actuelle et Questions restant en suspens ..................... 2
1.3. Décrire l’intérêt de mettre en place une telle étude ................................................ 2
1.4. Hypothèse de recherche ........................................................................................ 2
1.5. Retombées attendues de l’étude ............................................................................ 2
2. Objectifs ........................................................................................................................ 2
2.1. Objectif principal .................................................................................................... 2
2.2. Objectifs secondaires ............................................................................................. 2
3. Population et méthodes ................................................................................................. 3
3.1. Schéma d’étude ..................................................................................................... 3
3.2. Population étudiée ................................................................................................. 3
3.2.1. Description de la population ........................................................................... 3
3.2.2. Critères d’inclusion ......................................................................................... 3
3.2.3. Critères d’exclusion ........................................................................................ 3
3.2.4. Nombre de sujets nécessaires ....................................................................... 4
3.2.5. Mode et Faisabilité du recrutement ................................................................ 4
3.3. Randomisation ....................................................................................................... 4
4. Description de l’étude .................................................................................................... 4
4.1. Définition du moment d’inclusion dans la cohorte ................................................... 4
4.2. Constitution du groupe exposé ............................................................................... 4
4.3. Constitution du groupe non exposé ........................................................................ 4
4.4. Suivi ....................................................................................................................... 4
4.5. Critère de jugement ............................................................................................... 5
5. Déroulement pratique du protocole ................................................................................ 5
5.1. Visite de sélection .................................................................................................. 5
5.2. Organisation du suivi.............................................................................................. 5
6. Variables relevées ......................................................................................................... 5
6.1. Critères de jugement .............................................................................................. 6
6.2. Autres variables d’intérêts ...................................................................................... 6
6.3. Les sorties d’étude ................................................................................................. 6
7. Comité de suivi .............................................................................................................. 7
8. Circuit des données et Analyse statistique ..................................................................... 7
9. Ethique .......................................................................................................................... 7
10. Amendement et modification du protocole ................................................................. 8
11. Calendrier prévisionnel .............................................................................................. 8
12. Ressources nécessaires ............................................................................................ 8
13. Annexe 1 : Cahier d’observation ................................................................................ 9
14. Annexe 2 : Lettre d’information des patients et formulaire de consentement .............. 9
15. Annexe 3 : Engagement des directeurs d’Hôpitaux .................................................... 9
16. Annexe 4 : Lettre d’engagement des investigateurs ................................................... 9

__________________________________
Equipe de Méthodes en recherche clinique
Sandrine Andrieu, Catherine Arnaud, Thierry Lang, Valérie Lauwers-Cancès
2
1. Justification de l’étude
1.1. Définition de la maladie ou de l’événement étudié
Epidémiologie de la pathologie comportant fréquence et gravité, facteurs de risque,
conséquences individuelle et médico-économique.
1.2. Connaissance scientifique actuelle et questions restant en suspens
Présenter une synthèse récente de la bibliographie
1.3. Décrire l’intérêt de mettre en place une telle étude
Décrire les cohortes déjà existantes concernant cette pathologie, limites de la portée des
résultats (définition de la maladie différente, population différente, nombre insuffisant de
sujets, durée insuffisante de suivi…)
Intérêt et originalité d’une nouvelle étude.
1.4. Hypothèse de recherche
1.5. Retombées attendues de l’étude
2. Objectifs
2.1. Objectif principal
Il est unique et dépend du type de cohorte mise en place :
- soit on met en place une cohorte de sujets sains, diversement exposés à tel ou tel
facteur, que l’on suit pour voir apparaître une maladie et en étudier l’incidence (ou
un autre critère de jugement : une complication, le décès…)
- l’objectif sera d’étudier l’effet de l’exposition à un facteur donné sur la
survenue de la maladie
- soit on met en place une cohorte de patients (déjà atteints d’une pathologie
donnée) que l’on suit pour étudier l’histoire naturelle de la maladie, le pronostic de
la maladie
- l’objectif sera d’étudier les facteurs pronostics de la maladie (sur la survie,
la survenue de complications ou de récidives…)
2.2. Objectifs secondaires
Ils peuvent être multiples et dépendent du type de cohorte mise en place.

__________________________________
Equipe de Méthodes en recherche clinique
Sandrine Andrieu, Catherine Arnaud, Thierry Lang, Valérie Lauwers-Cancès
3
3. Population et méthodes
3.1. Schéma d’étude
Etude de cohorte, préciser :
- multicentrique ou monocentrique,
- fixe ou fermée (toutes les inclusions se font au début de l’étude) ou ouverte (les
inclusions pourront se poursuivre à l’avenir au fur et à mesure que les sujets
présenteront les critères d’inclusion) ,
- contemporaine (l’inclusion des sujets se fait au début de l’étude) ou historique (les
sujets sont déjà inclus quand l’étude débute et on tente de reconstruire à
posteriori la cohorte)
3.2. Population étudiée
3.2.1. Description de la population
- sujets sains ou malades
- nombre de sujets dans chaque groupe (que l’on justifie ensuite)
3.2.2. Critères d’inclusion
- soit on met en place une cohorte de sujets sains, diversement exposés à tel ou tel
facteur de risque, que l’on suit pour voir apparaître une maladie et en étudier
l’incidence (ou un autre critère de jugement : une complication, le décès…)
- définir précisément l’exposition au facteur étudié
- soit on met en place une cohorte de patients (déjà atteints d’une pathologie, que
l’on suit pour étudier le pronostic de la maladie
- définir précisément les critères diagnostiques de la pathologie
Dans tous les cas :
- la possibilité d’un suivi pour le temps de suivi qui a été prédéfini
- patient volontaire (à voir : ayant signé le consentement éclairé)
3.2.3. Critères d’exclusion
- soit on met en place une cohorte de sujets sains, diversement exposés à tel ou tel
facteur de risque, que l’on suit pour voir apparaître une maladie et en étudier
l’incidence (ou un autre critère de jugement : une complication, le décès…)
- les personnes atteintes de la maladie étudiée ou du problème de santé
- soit on met en place une cohorte de patients (déjà atteints d’une pathologie),
diversement pris en charge, que l’on suit pour étudier le pronostic de la maladie.
- les personnes porteuses d’une complication de la maladie étudiée…
Dans tous les cas : Absence de suivi possible

__________________________________
Equipe de Méthodes en recherche clinique
Sandrine Andrieu, Catherine Arnaud, Thierry Lang, Valérie Lauwers-Cancès
4
3.2.4. Nombre de sujets nécessaires
Pour un test bilatéral, un risque d’erreur de 0.05 et une puissance de l’étude de 90%, afin
de mettre en évidence une différence au moins égale à X% entre les deux groupes à l’issue
du suivi, le nombre de sujets à recruter est de X. Ce nombre est majoré de XX% pour tenir
compte des perdus de vue.
Cette estimation peut être effectuée à partir de la page Web suivante :
Interactive Statistical Calculation Pages
Ce calcul peut s’avérer beaucoup plus compliqué, notamment si le suivi des sujets n’est pas
identique pour tous. Il est à déterminer avec une personne de l’équipe « méthodes en
recherche clinique » du Service d’Epidémiologie.
3.2.5. Mode et Faisabilité du recrutement
Le recrutement se fera à partir des services de XX et de XX participants à l'étude
(précisez si les sujets sont recrutés par la consultation, l’hospitalisation, ou une autre source)
et capables de mettre en œuvre un suivi. La participation de plusieurs centres est acquise :
lister les centres. Le nombre moyen de patients par an ou par mois dans chaque centre
étant de X… , la durée des inclusions devrait être de X….
3.3. Randomisation
Certaines études de cohortes peuvent faire l’objet d’une randomisation (un essai
thérapeutique est une étude de cohorte particulière).
Cf chapitre Randomisation de l’essai thérapeutique
4. Description de l’étude
4.1. Définition du moment d’inclusion dans la cohorte
Définir précisément le temps zéro qui correspond au début du suivi (installation d’un
symptôme, diagnostic d’une pathologie, âge seuil…).
4.2. Constitution du groupe exposé
Définir précisément l’exposition : nature, niveau, ancienneté, minimum d’exposition
4.3. Constitution du groupe non exposé
Définir les critères de choix des sujets non exposés : absence d’exposition ou faible niveau
d’exposition.
Ce groupe peut faire partie de la cohorte ou être externe à la cohorte.
4.4. Suivi
Décrire la procédure de suivi pour les deux groupes tout au long de l’étude :
- modalité de suivi (visites, courriers, alternance)
- périodicité des contacts
- durée totale du suivi

__________________________________
Equipe de Méthodes en recherche clinique
Sandrine Andrieu, Catherine Arnaud, Thierry Lang, Valérie Lauwers-Cancès
5
4.5. Critère de jugement
Définir précisément l’événement recherché et préciser si ce critère est validé (joindre une
référence bibliographique) et reproductible :
- Apparition de la maladie
- Décès
- Réponse à un traitement (définir le type de critère : biologique, radiologique ou
clinique…)
- Rémission (définir le type de critère : biologique, radiologique ou clinique…)
- Récidive (définir le type de critère : biologique, radiologique ou clinique…)
- Qualité de vie
5. Déroulement pratique du protocole
Il faut décrire en pratique, pour un patient donné, les différents temps du protocole.
5.1. Visite de sélection
Elle intervient après la vérification des critères d’inclusion et d’exclusion des patients. A la fin
de cette visite, le consentement écrit du patient est obtenu.
Décrire qui sera responsable de cette visite, où elle aura lieu et quels sont les examens
complémentaires qui seront effectués.
5.2. Organisation du suivi
Les patients seront suivis aux temps suivants ………………….
Décrire et justifier les différents temps du suivi, qui sera en charge de celui ci et quels sont
les examens qui seront faits à tous les patients.
Comment sera organisé le suivi.
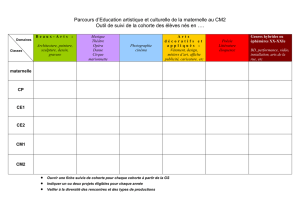
Faire un tableau récapitulatif des différents examens qui seront réalisés durant le suivi.
6. Variables relevées
Le recueil des données sera effectué à l'aide de cahiers d'observation standardisés
qui comprendront les caractéristiques des patients, des facteurs d’exposition, de la
pathologie étudiée et les données du suivi. Ce cahier figure en annexe.
Le recueil sera centralisé, les cahiers d'observation seront retournés à chaque temps
d'observation dans le service de mentionner la spécialité du centre coordinateur. Chaque
investigateur devra compléter et signer les feuillets du cahier d'observation qui le concernent.
Ces cahiers devront être complétés de façon lisible au stylo à bille, chaque correction
effectuée devant être datée, et signée par le médecin, la donnée initiale devant rester visible.
La vérification de la qualité des données sera effectuée par l'assistant de recherche
clinique affecté à cette étude, qui saisira dès réception les données et recherchera
l'existence de données manquantes et aberrantes. Toutes données ainsi retrouvées, feront
l'objet d'une demande de correction par l’investigateur du centre ayant adressé le cahier
d'observation.
 6
7
8
9
6
7
8
9
1
/
9
100%