Compte rendu des IIIèmes Journées SFM, Société Française de

© AFM - Institut de Myologie 2005 - -
La reproduction des textes est soumise à l’appréciation de l’AFM et des auteurs
1
Compte rendu des IIIèmes Journées SFM,
Société Française de Myologie
20 & 21 octobre 2005 à Paris
Les IIIèmes Journées Annuelles de la Société Française de Myologie se sont déroulées à
l’Institut de Myologie à Paris.
Cette troisième édition consacrée au thème « jonction neuromusculaire, biologie et
pathologie » et organisée cette année par Daniel Hantaï sous la présidence de Michel
Fardeau, a réuni environ 140 médecins et chercheurs. La manifestation a débuté le jeudi 20
octobre par le groupe d’étude en myologie suivi d’une conférence donnée par Jacques Taxi
en l’honneur du Professeur René Couteaux, éminent spécialiste de la jonction
neuromusculaire. La deuxième journée s’est déroulée en trois temps : biologie de la
jonction neuromusculaire, syndromes myasthéniques congénitaux et myasthénie
autoimmune.
Biologie de la jonction Neuromusculaire
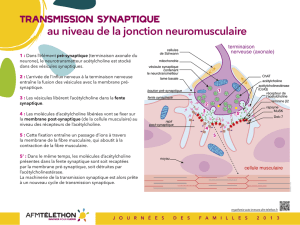
Au niveau de la jonction neuromusculaire, l'acétylcholine (ACh), neuromédiateur stocké
dans les vésicules synaptiques, est massivement libérée au moment de l’arrivée de
l’influx nerveux du motoneurone. L’ACh libérée traverse la lame basale et se fixe sur
les récepteurs à l'acétylcholine (RACh), qui s’ouvrent de manière transitoire. Lorsque la
dépolarisation de la membrane post synaptique atteint le seuil d’activation des canaux
sodiques du muscle, le potentiel d’action se propage alors à la surface de la fibre. C’est
la transmission synaptique.
La libération de calcium induite par cette dépolarisation provoque la contraction de la
fibre. Dans la fente synaptique, l'acétylcholine est rapidement dégradée par
l'acétylcholinestérase (AChE) permettant ainsi une contraction ultérieure.
Communication d’Eric Krejci : Cholinestérase et contrôle de la transmission
synaptique à la jonction neuromusculaire
Chez les mammifères, l’acétylcholine peut être hydrolysée par deux enzymes :
l'acétylcholinestérase (AChE) et la butyrylcholinestérase (BChE). Ces deux enzymes
possèdent des caractéristiques catalytiques différentes mais des mécanismes
moléculaires communs d’assemblage en oligomères.
Les travaux d’Eric Krejci ont montré que deux protéines, PRiMA et ColQ, sont capables
d’agencer des complexes tétramériques d’AChE et de BChE et de les adresser dans
deux compartiments de la jonction neuromusculaire : dans les lames basales post-
synaptiques via ColQ et dans les membranes plasmiques via PRiMA.
Des manipulations génétiques chez la souris ont permis de supprimer le domaine
d’interaction (PRAD) de ColQ et de PRiMA avec les cholinestérases (entraînant une
absence des complexes) et d’en étudier les conséquences sur la localisation de l’AChE
dans la jonction neuromusculaire.
En absence de ColQ (modèle de myasthénie congénitale), le niveau d’AChE est très
faible et les RACh sont activés de manière répétitive. La BChE qui est toujours
accumulée dans ce mutant ne contrôle pas la durée d’activation des récepteurs mais
modulent les processus de libération de l’ACh. Ainsi, l’excès d’acétylcholine entraîne
non seulement des modulations post-synaptiques mais aussi des modifications
présynaptiques.

© AFM - Institut de Myologie 2005 - -
La reproduction des textes est soumise à l’appréciation de l’AFM et des auteurs
2
Communication de Laurent Schaeffer : Contrôle de l’expression des gènes
musculaires par l’innervation motrice
Dans le muscle adulte, la majorité des protéines ayant une fonction synaptique
(comme le récepteur à l’acétylcholine) est spécifiquement localisée sous la terminaison
nerveuse. En effet, les gènes codant ces protéines (appelés gènes synaptiques) sont
spécifiquement exprimés par quelques noyaux situés directement sous la terminaison
nerveuse (noyaux sous-neuraux) alors qu’ils ne sont pas exprimés dans les noyaux
situés plus loin de la jonction neuromusculaire (noyaux extra-synaptiques). Cette
compartimentation de l’expression des gènes résulte de l’action conjointe de deux
mécanismes régulateurs contrôlés par l’innervation :
- Dans les noyaux sous-neuraux, les facteurs agrine er neurégulines activent les
récepteurs MusK, déclenchant une cascade de réactions moléculaires (voie MAPK)
aboutissant à la stimulation de l’expression des gènes synaptiques via le facteur de
transcription GABP.
- Dans les régions extra-synaptiques, l’activité électrique (due à la transmission
nerveuse) réprime l’expression de gènes, en particulier RACh, via une voie de
signalisation calcique.
D’après les travaux de Laurent Schaeffer, ces deux mécanismes impliquent le contrôle
de l’état de compaction de la chromatine* dans les noyaux musculaires sous-neuraux
et extra-synaptiques. En effet, lorsque l’ADN est condensé, il est moins accessible aux
facteurs de transcription et l’expression des gènes est inhibée. Par contre, lorsque
l’ADN est relâché, les gènes peuvent être transcrits.
*Dans les cellules eucaryotes, le matériel génétique est organisé en une structure
complexe constituée d'ADN et de protéines, localisé dans le noyau. Cette structure a
été baptisée chromatine (du grec khroma: couleur et sôma: corps). La chromatine peut
être plus ou moins compactée.
Syndromes myasthéniques congénitaux (SMC)
Les syndromes myasthéniques congénitaux (SMC) constituent un groupe hétérogène
de maladies génétiques affectant la transmission neuromusculaire au niveau :
pré-synaptique : mutations du gène CHAT (choline acétyltransférase);
synaptique : mutations du gène COLQ (queue collagénique de
l'acétylcholinestérase) entraînant un déficit en AChE;
post-synaptique : mutations des gènes CHRNA1, CHRNAB1, CHRND, CHRNE
(sous-unités du récepteur de l’acétylcholine), RAPSN (rapsyne), SCN4A (canal
sodium musculaire), MUSK (récepteur tyrosine kinase musculaire).
La caractérisation des SMC comprend deux étapes : établir le diagnostic clinique puis
identifier et caractériser l’anomalie moléculaire.
Daniel Hantaï :
Réseau français sur les syndromes myasthéniques congénitaux
Les SMC sont des maladies héréditaires chroniques, rares et invalidantes, débutant
souvent dès l'enfance et pouvant mettre en jeu le pronostic vital. Ils ont en commun
une anomalie de la transmission neuromusculaire qui s’exprime cliniquement par une
faiblesse musculaire accentuée par l’effort. Ils forment cependant un groupe
d’affections hétérogènes tant sur le plan clinique, de l’hérédité et des mécanismes
physiopathologiques qui les sous-tendent ce qui explique la difficulté d'un diagnostic
physiopathologique complet.
Nous avons mis sur pied en 2001 un réseau clinique et de recherche sur les SMC. Il
réunit des services cliniques de neuropédiatrie et de neurologie adulte et des
laboratoires de recherche spécialisés dans l'étude la jonction neuromusculaire
(tableau). Il a permis de développer pour la première fois en France l’approche et les
moyens nécessaires à une caractérisation des SMC qui repose sur la clinique,
l’électroneuromyographie, l’étude des jonctions neuromusculaire sur biopsie musculaire

© AFM - Institut de Myologie 2005 - -
La reproduction des textes est soumise à l’appréciation de l’AFM et des auteurs
3
et l’identification de mutations dans les 9 gènes connus à ce jour, le dernier venant
d’être identifié par notre réseau.C’est ainsi qu’ont pu être étudiés les 100 patients
recensés alors dans la population française et une solution moléculaire a pu être
donnée pour 55 d'entre eux permettant un traitement adapté et, pour certains, un
conseil génétique.
Outre l'identification de mutations dans les 9 gènes connus, nous recherchons de
nouveaux gènes responsables des SMC dans l'ensemble des patients non caractérisés
actuellement. Ceci passe par une stratégie classique d'analyse liaison quand elle
s'avère possible mais surtout par une stratégie gène candidat qui repose sur l’étude en
parallèle des biopsies musculaires des patients et de leur ADN. Pour démontrer le
caractère pathogène des mutations nouvelles identifiées mais aussi dans la perspective
de nouvelles thérapeutiques, nous utilisons et développons des modèles d’expression
de ces mutations (modèles cellulaires et modèles murins).
Ce réseau s'appuie sur les centres de référence en pathologie neuromusculaire
nouvellement labellisés et réunit des équipes cliniques et des groupes de recherche de
très haut niveau. Il permet tout à la fois de développer les outils diagnostiques et les
modèles expérimentaux seuls à même de parvenir à une caractérisation complète des
SMC afin d’autoriser un traitement adapté et de donner un conseil génétique approprié
aux patients.
EQUIPES CLINIQUES
PARIS
Bruno Eymard, Michel Fardeau,
Pitié-Salpêtrière
Pascale Richard, Pitié-Salpêtrière
Michèle Mayer, St Vincent de
Paul
GARCHES
Brigitte Estournet, Raymond
Poincaré
Bernard Clair, Raymond Poincaré
LE PLESSIS-ROBINSON
Philippe Dartevelle, Marie-
Lannelongue
LILLE
Tanya Stojkovic, Roger Salengro
LYON
Guy Chauplannaz, Hôpital
Neurologique
MARSEILLE
Jean Pouget, La Timone
MONTPELLIER
Bernard Echenne, St Eloi
NICE
Claude Desnuelle, L’Archet
STRASBOURG
Christine Tranchant, Hôpitaux
Universitaires
TUNIS
Fayçal Hentati, La Rabta
TOKYO
Makiko Osawa, Tokyo’s Women
University Hospital
GROUPES DE RECHERCHE
PARIS
Daniel Hantaï, INSERM U582,
Pitié-Salpêtrière
Eric Krejci, Claire Legay, ENS
Paris
Pierre Jean Corringer, CNRS URA
2182, Institut Pasteur
GIF sur YVETTE
Jordi Molgo, CNRS UPR 9040
Takeshi Shimahara, CNRS UPR
9040
LYON
Laurent Schaeffer, ENS/CNRS
UMR 5665
HEIDELBERG
Veit Witzemann, Max Planck
Institut
VIENNE
Ruth Herbst, Wien Universität

© AFM - Institut de Myologie 2005 - -
La reproduction des textes est soumise à l’appréciation de l’AFM et des auteurs
4
Communication d’Emmanuel Fournier : Diagnostic EMG des syndromes
myasthéniques congénitaux
L’objectif de l’électromyographie est de mettre en évidence les anomalies de la
transmission nerveuse et de les caractériser afin d’orienter le diagnostic moléculaire et
de guider le choix thérapeutique. Certains signes caractéristiques à l’EMG sont
évocateurs d’un certain type de SMC : les réponses motrices répétitives sont le signe
d’une SMC avec excès de fonction de la jonction neuromusculaire (contre indication
aux anti-cholinestérasiques) alors qu’un décrément isolé à 3Hz (sans dédoublement
des réponses motrices) évoque plutôt un phénotype de défaut de fonction de la
jonction neuromusculaire (orientation vers un traitement anti-cholinestérasique).
L’étude électromyographique a porté sur 77 patients examinés pour suspicion de
syndrome myasthénique congénital (SMC).
Des réponses motrices répétitives ont été caractérisées chez 9 d’entre eux. L’analyse
moléculaire a révélé que 4 de ces patients étaient porteurs de mutations du gène COLQ
(déficit en cholinestérase) et 3 de mutations responsables de syndrome du canal lent,
ce qui correspond effectivement à un gain de fonction de la JNM. Le diagnostic
génétique a donc confirmé à 78% des résultats prédictifs de l’EMG.
Chez 46 autres patients, l’EMG a mis en évidence un décrément à la stimulation
répétitive 3 Hz, sans dédoublement des réponses motrices, orientant vers un défaut de
la jonction neuromusculaire. L’analyse moléculaire a confirmé à 45% ces prédictions :
mutations responsables de déficits en récepteur à l’acétylcholine (10 patients), rapsyne
(9 patients) et MuSK (1 patient).
Chez un certain nombre de patients souffrant essentiellement d’une faiblesse
musculaire proximale (soit une présentation plus myopathique que myasthénique),
l’examen EMG a révélé des anomalies de transmission neuromusculaire insoupçonnées.
D’après ces résultats, un des objectifs de l’EMG serait de rechercher un SMC chez des
cas myopathiques sans explication évidente.
Signes à l’EMG
Réponses motrices
répétitives
Décrément isolé à 3Hz
(sans dédoublement
des réponses
motrices)
Type de SMC
évoqué
SMC avec excès de
fonction de la jonction
neuromusculaire
SMC défaut de
fonction de la jonction
neuromusculaire
Orientation du
traitement
Contre indication aux
anti-
cholinestérasiques
Anti-
cholinestérasiques
Communication de Pascale Richard : Génétique des Syndromes
Myasthéniques Congénitaux (SMC) : de la mutation privée à l’effet fondateur
Les études génétiques dans les syndromes myasthéniques sont récentes et ont déjà
montré l’existence d’une importante hétérogénéité génétique.
- Très peu de mutations du gène CHAT codant la Choline Acétyl transférase ont été
identifiées dans les formes présynaptiques.
- Dans les formes synaptiques avec déficit en acétylcholine, des mutations du gène
COLQ codant la queue collagénique de l’acétylcholinestérase sont retrouvées
fréquement.
Dans les SMC postsynaptiques, 20% des patients sont mutés dans CHRNE (sous unité
epsilon du récepteur de l’acétylcholine) et 20% dans RAPSN (rapsyne).
De très nombreuses mutations sont décrites dans ces gènes et dans la grande majorité
des cas, chaque famille ou patient présente sa propre mutation, suggérant qu’il s’agit
de « mutations privées ». Cependant, quelques mutations sont retrouvées avec une
plus grande fréquence dans certaines populations, et montrent l’existence d’un effet

© AFM - Institut de Myologie 2005 - -
La reproduction des textes est soumise à l’appréciation de l’AFM et des auteurs
5
fondateur. Il s’agit entre autre de la mutation I336T de CHAT dans des familles
d’origine turque, la mutation G240X de COLQ qui aurait un effet fondateur au Moyen
Orient, la mutation gitane 1267delG dans CHRNE et la mutation N88K dans RAPSN qui
a un effet fondateur d’origine indo-européenne.
Pascale Richard et son équipe ont montré que la mutation 1293insG de CHRNE est très
souvent présente chez les patients originaires du pourtour méditerranéen (60%) par
rapport à l’ensemble des mutations de ce gène chez des patients Européens (20%).
L’analyse d’une cohorte de patients porteurs de cette mutation a montré l’existence
d’un effet fondateur dans ces populations.
Dans un objectif diagnostique, la recherche directe de cette mutation chez des patients
issus du pourtour méditerranéen permet une identification rapide de l’anomalie
moléculaire en cause.
Haut de page
Communication de Thierry Kuntzer : Aspects phénotypiques d’un syndrome
myasthénique congénital (SMC) dû à des mutations du gène COLQ
Thierry Kuntzer a discuté les manifestations cliniques et électrophysiologiques d’un
patient de 45 ans ayant un syndrome myasthénique congénital s'exprimant par une
parésie appendiculaire asymétrique et respiratoire, et s’étant révélé porteur d’une
double mutation hétérozygote du gène COLQ; une amélioration motrice par la prise
d’éphédrine est espérée.
Par ailleurs, des observations cliniques de patients présentant un SMC ont été
présentées par Thierry Kuntzer et Tanya Stojkovic. L'effet bénéfique apparemment
paradoxal de la 3,4-diaminopyridine chez les patients présentant une mutation dans le
gène COLQ a été discuté.
<< retour
Myasthénie autoimmune
La myasthénie autoimmune est une maladie multifactorielle. La plupart des sujets
atteints de myasthénie autoimmune (plus de 80%) fabriquent des auto-anticorps se
fixant sur les récepteurs de l'acétylcholine. En conséquence l'acétylcholine ne peut plus
se fixer au niveau de la membrane musculaire et la commande ne passe plus du nerf
vers le muscle. Parmi les personnes qui ne présentent pas d'anticorps anti-RACh, 41%
ont des anticorps anti-MuSK. MuSK est un récepteur tyrosine-kinase spécifique du
muscle, qui joue un rôle important dans le développement et la stabilité de la
membrane musculaire. Les personnes atteintes de myasthénie avec anticorps anti-
MuSK se différencient des autres par la résistance au traitement par anti-
cholinestérasiques. Il existe enfin des patients atteints de myasthénie sans anticorps
anti-RACh et sans anticorps anti-MuSK.
Communication de Henri-Jean Garchon : Association génétique de la sous-
unité alpha du récepteur à l’acétylcholine dans la myasthénie autoimmune
Bien que la myasthénie autoimmune ne soit pas une maladie héréditaire, le rôle de
facteurs génétiques prédisposants ou au contraire protecteurs est maintenant bien
reconnu. Leur étude est toutefois complexe. En effet, d’une part, il s’agit en général de
polymorphismes communs qui sont également présents dans la population générale,
chez des personnes non malades. D’autre part, un phénomène génétique très répandu,
appelé déséquilibre de liaison, qui consiste en l’association préférentielle d’allèles de
gènes voisins, empêche de discriminer aisément les effets de locus proches.
L’association du complexe HLA dans la forme de myasthénie la plus fréquente, celle
touchant des femmes jeunes et associée à une hyperplasie du thymus, est connue
depuis longtemps. Le travail récent de Claire Vandiedonck (équipe de H-J Garchon) a
démontré pour la première fois, grâce à une analyse avec une première carte de
marqueurs de résolution intermédiaire (1 marqueur pour 100 000 bases), la complexité
 6
7
8
6
7
8
1
/
8
100%