Cours du 11-03 - Ronéos master M1 santé d`immunologie

1
Master Immunologie Aymeric Becq
Cours du 11/03/08 Nicolas de Chanaud
Déficits Immunitaires Héréditaires
Plan (à titre indicatif):
Introduction
I. Immunitée adaptative, le développement lymphocytaire
a. SCID avec absence de LT et NK
i. Forme récessive liée à l’X
ii. Forme autosomique récessive
b. SCID avec absence de LB et LT
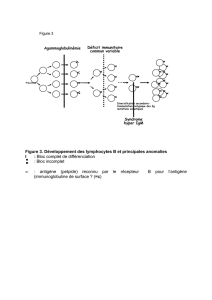
c. Défaut de commutation isotypique
II. Immunitée anti-infectieuse
III. Conséquence sur l’auto-immunité
a. Défaut de sélection négative :
b. Défaut contrôle de la vie et de la mort des Lb et Lt.
Introduction:
Les déficits immunitaire héréditaires sont des maladies que l’on rencontre chez
l’homme (mais aussi chez certains animaux) avec une faible fréquence de l’ordre de 150
cas sur 800.000 naissances. Ces maladies ne sont donc pas un problème médical majeur
(en termes de santé public bien sur).
Chacune de ces maladies est un modèle qui nous permet d’étudier le système
immunitaire.
En effet, pour la grande majorité d’entres elles, il s’agit d’une mutation génique (maladie
héréditaire monogénique de type mendélienne). On peut donc analyser la fonction de la
protéine (codée par ce gène) qui est ici déficiente.
Aujourd’hui, on connait environ 150 de ces maladies (ce qui fait autant de modèles à
étudier). Un gène muté peut être associé à 130 d’entre elles minimum ce qui permet de
déduire la physiopathologie de la maladie et, par voie de conséquence, la physiologie
normale (soit : à quoi sert la protéine ?).
Ces anomalies touchent tous les aspects de l’immunité: l’immunité innée et adaptative, le
développement, la différenciation des différentes cellules qui interviennent dans la
réponse immunitaire, les fonctions effectrices des 2 types d’immunités, la régulation de la
réponse immunitaire.
Ces maladies sont aussi une manière d’étudier chez l’homme quels sont les effecteurs de
l’immunité infectieuse.
On peut associer une mutation, à une susceptibilité des patients face à tel ou tel type de
micro-organisme et en déduire quel a pu être le rôle de la protéine, dans un type
d’immunité, face à un type de micro-organisme donné.

2
Parmi les conséquences possibles de ces maladies on compte le développement de
maladies auto immunes qui sont des modèles particuliers et rares. Ces dernières sont une
manière d’étudier les systèmes de contrôle des réponses auto réactives.
I. Immunité adaptative: le développement lymphocytaire.
On aborde ici des pathologies très rares (10 naissances par an en France) qui sont
les Déficits immunitaires combinés sévères (SCID).
Pour cela nous disposons d’un modèle naturel (non provoqué) chez la souris appelée
souris SCID.
Ces maladies fournissent des informations sur le développement lymphocytaire,
notamment des lymphocytes T (LT).
Ces maladies sont un défaut complet de différenciation des LT en raison d’une anomalie
génétique héréditaire intrinsèque de l’hématopoïèse (lymphopoïèse ici).
Les enfants naissent sans LT (sans organes lymphoïdes périphériques), il n’y a ni LT ni
précurseurs de LT dans le thymus. C’est une anomalie propre à la lignée des LT et non
une conséquence de l’environnement où ces derniers se différencient.
Une quinzaine de mutations géniques différentes ont été dénombrées à ce jour, qui sont
responsables de la maladie. Les protéines codées par ces gènes jouent donc un rôle dans
la différenciation des LT.
Ces maladies sont classées en fonction du type de défaut de différenciation
lymphocytaire.
-Dans quelques cas il y existe un défaut de développement des LT unique c’est à
dire sans autre anomalie associée.
-On peut être en présence d’un défaut de développement des LT et LB (les autres
lignées sont épargnées). Le gène muté codé pour une protéine joue un rôle dans le
développement des LT et des LB.
- Il peut également y avoir un défaut de développement des LT et des cellules NK
(Natural Killer).
- Enfin, exceptionnellement, il peut exister un défaut de la lymphopoïèse
(lymphocytes T, B, et NK) et de la myélopoïèse (PNN: polynucléaires neutrophiles).
Auquel cas, il s’agit d’une maladie encore plus sévère puisqu’elle touche l’immunité
adaptative et innée.
L’apparition de l’immunité adaptative date d’il y a 450 millions d’années. Cette
immunité est absente chez les insectes notamment (toute espèce étant d’apparition plus
ancienne que 450 MA). Ces derniers n’ont donc pas de déficit immunitaire.
En revanche, l’homme qui nait avec ce type de déficit : sans LT, par exemple, décède-en
l’absence de traitement- de toutes sortes de maladies infectieuses provoquées par des
germes pathogènes (touchant aussi des sujets normaux) ou par des germes opportunistes
(non pathogènes pour un sujet normal) en l’espace de quelques mois. L’espérance de vie
est donc très réduite.
L’immunité adaptative (LT…) est donc essentielle pour la longévité chez l’homme.

3
Cela explique partiellement la longévité limitée de toutes les espèces vivantes dépourvues
d’immunité adaptative (sauf exceptions, notamment dans le monde végétal).
Deux exemples de SCID seront étudiés à présent:
a. SCID avec absence de LT et NK (3éme catégorie):
Deux types de transmissions génétiques sont à distinguer:
i. Forme récessive liée à l’X (la plus fréquente):
On a pu localiser la région du chromosome X où se trouve le gène muté.
Le gène codant pour la chaîne gamma du récepteur de l’Interleukine2 (IL2) a été
découvert plus tard dans la même région que celle où se trouve le gène muté cité ci-
dessus.
Dés lors, il est possible de supposer que ce gène codant pour la chaine gamma est muté
dans la maladie puisqu’il se situe à proximité du gène qui muté provoque la maladie.
Ce raisonnement est pourtant immunologiquement faux, à l’époque, car l’IL2 n’est pas
impliqué dans le développement des LT.
Des recherches ont plus tard prouvé que ce gène est effectivement muté chez les
malades. Cela parait pourtant incohérent.
L’hypothèse suivante fut alors formulée: cette chaine gamma doit avoir d’autres
fonctions qui expliqueraient le trouble de développement des LT observé.
On a progressivement montré que la chaine gamma est associée à 6 récepteurs de
cytokine. Elle joue donc un rôle majeur dans la signalisation pour un grand nombre de
cytokines (IL 2,4,7,9,15,21).
On l’a appelée la chaine gamma commune: gamma c
Par la suite, il s’est agit de savoir quel(s) récepteur(s) joue(nt) un rôle dans le
développement des LT et NK. On savait simplement que ce n’était pas le cas pour l’IL2.
On a démontré que les enfants atteints d’un SCID avec absence de LT (1ère catégorie)
étaient porteurs d’une mutation d’un gène codant pour la chaine alpha du récepteur de IL
7. Cette cytokine joue donc un rôle majeur dans le développement des LT car sa
déficience entraine un défaut complet de développement des LT. L’IL 7 transmet via son
récepteur, des signaux de survie et de prolifération aux précurseurs de LT au stade de
double négatif, c’est à dire avant le réarrangement des gènes codant pour le TCR, durant
la phase de prolifération.
La chaine gamma c du récepteur de IL15 est nécessaire au développement des NK.
Ainsi il est devenu possible de cerner, (en 20 ans!), en observant les malades, le rôle les
différentes cytokines dans le développement lymphocytaire.
ii. Forme autosomique récessive:
Le gène muté ici, provoque exactement la même maladie. Il code pour la kinase JAK3.
JAK3 est capable de phosphoryler des tyrosines. Cette kinase s’associe au domaine
intracellulaire de la chaine gamma c (normale ici!) du récepteur de l’IL7.
Quand IL7 interagit avec son récepteur, cela active JAK3 qui phosphoryle ses propres
résidus tyrosine ainsi que ceux sur gamma c, ce qui permet le recrutement d’autres
protéines, les protéines STAT.

4
Ces facteurs de transmission se dimérisent et migrent dans le noyau pour activer la
transcription d’un certain nombre de gènes.
Quand JAK3 est absente, la signalisation est bloquée (même si le récepteur est normal).
Schéma 1:
Exemple:
Nous allons étudier à présent, un cas particulier car, très rare.
Schéma 2:
L’oncle maternel de l’enfant auquel nous nous intéressons est mort d’un SCID après 1 an
de vie. La chaine gamma c étant mutée chez cet oncle, il y avait une absence de LT et
NK.
L’enfant quand à lui, présente des infections modérées à un an de vie. En étudiant ses
globules blancs, on s’est aperçus qu’il possède des LT (50% par rapport à la normale)
mais pas de NK. Il n’a donc pas la même maladie que son oncle.
Sa mère est conductrice (1 allèle muté) de sorte qu’il a hérité de la mutation gamma c…
mais comment expliquer qu’il possède des LT??
D’aucuns pourraient penser que ces LT appartiennent à sa mère (transfusion naturelle à
la naissance) et qu’ils persistent en absence de réaction immunitaire de l’enfant. Mais ce
n’est pas le cas comme l’ont prouvé des tests non détaillés ici.
En effet, on s’est s’aperçu que les LT de l’enfant expriment la chaine gamma c
normalement !!
père
fils
mère
Oncle
décédé
R à IL-7
IL-7
α
γc
JAK3
STAT

5
Remarque: attention!! Il a tout de même hérité de la mutation, donc toutes les autres
cellules de cet enfant portent cette mutation.
Explication: Il y a eu une mutation somatique qui a corrigée l’erreur à l’endroit même où
se situait la mutation germinale d’origine. Ce type d’évènement est bien sûr très rare. Les
LT de l’enfant sont donc issus du précurseur où la mutation somatique a eu lieu.
Schéma 3:
Dans la moelle osseuse de l’enfant, il y a des précurseurs de LT tous porteurs du
gène muté.
Puis dans l’un d’entre eux, l’évènement de mutation somatique survient. Cette cellule va
migrer vers le thymus (grâce à son récepteur normal) pour s’y diviser puis se différencier.
On s’intéresse alors à la diversité du répertoire des LT, c’est à dire combien de clones de
LT existent en tant que récepteurs T pour l’antigène.
Cela permettra de mesurer la capacité intrinsèque d’un précurseur de LT à générer un
répertoire (combien de cellules différentes?).
Tout dépend de la phase de prolifération qui précède le réarrangement et détermine le
nombre de clones qui ensuite vont se différencier par rapport à ces réarrangements.
On observe la diversité de la chaine Béta du TCR:
Parmi les clones de LT mémoires (pas de LT naïfs) analysés dans le sang de l’enfant, on
en recense 1000 différents (pour 100 000 normalement).
Il s’agit là d’une sous estimation car on n’étudie pas les organes lymphoïdes de l’enfant
(seulement son sang).
Cet enfant a donc 1% de la diversité de la chaîne Beta du TCR, générée par une seule
cellule.
C’est ce qui lui a permis de vivre plusieurs années et de survivre à plusieurs infections
(notamment une varicelle).
Une cellule est donc capable de générer 1% du répertoire complet (1000 clones différents
pour la chaine Bêta, sans prendre en compte, en ce cas, la combinatoire avec la chaine
Alpha du TCR).
On calcule que pour passer de 1 à 1000 cellules, il faut environ 10 divisions cellulaires
d’où la possibilité de déterminer que la phase de prolifération, qui dépend de l’IL7, est
considérable car les cellules se sont multipliées au moins dix fois.
 6
7
8
9
10
11
6
7
8
9
10
11
1
/
11
100%