Diagnostic anténatal en génétique moléculaire

Diagnostic anténatal en génétique moléculaire
1. Détection directe de la mutation
Anémie falciforme
Nous avons déjà vu (Ch3) que la cause de l’anémie falciforme est une mutation qui conduit au
remplacement d’un acide glutamique par une valine en position 6 le la globine (exprimée
uniquement dans l’âge « adulte »).
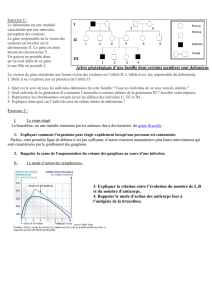
Figure 8- 1. Séquence des deux allèles et des oligonucléotides utilisés pour la détection de
l'anémie.
Cette unique différence de séquence entre les deux allèles est utilisée pour détecter quels sont
ceux portés par un sujet donné. Des séquences exactement complémentaires d’une petite
portion de la région du gène qui porte la modification ont été synthétisées in vitro (figure 8-1).
On a mis au point un protocole expérimental qui permet d’hybrider les sondes
oligonucléotidiques uniquement lorsque cette hybridation est exacte (aucun mésappariement).
Un sujet homozygote sain ne porte que l’allèle b+ et son ADN n’hybride qu’avec la sonde 1,
un sujet malade ne porte qu’un allèle ßS et son ADN n’hybride qu’avec la sonde 2, quant à un
sujet sain mais porteur (hétérozygote) il porte les deux allèles et son ADN hybride avec les
deux sondes. Pour un diagnostic prénatal, on prélève des cellules amniotiques (d’origine
fœtale) que l’on cultive pour en extraire l’ADN qui est ensuite hybridé séparément avec
chaque sonde marquée.
Figure 8- 2. Détection de la forme allélique conférant l'anémie falciforme.

Chorée de Huntington
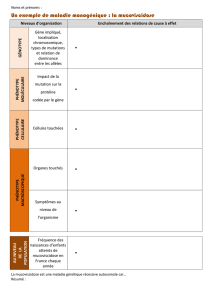
Il s'agit d'une maladie neurodégénérative qui se déclare en général entre 35 et 50 ans. Des
altérations physiques et mentales peuvent apparaître simultanément, venir l'une après l'autre
ou encore de manière alternative.
* parmi les modifications physiques, on note l'agitation, l'apparition de mouvements
désordonnés, des difficultés d'élocution et de déglutition.
* Les troubles mentaux se manifestent par des modifications de la personnalité avec, par
exemple, une irascibilité permanente, une apathie, un état dépressif, une perte de volonté, un
repli sur soi.
Figure 8- 3. Age où la maladie se déclare chez un sujet porteur d'un allèle défectueux.
Figure 8- 4. Pedigree d'une famille présentant des membres atteints de chorée de
Huntington.

Figure 8- 5. Diagnostic prénatal de la Chorée de Huntington.
La transmission de cette maladie est de type dominant, autosomique (un père II-6 a des fils
atteints). Le gène responsable de la maladie a été localisé sur le chromosome 4 en 1983 et
cloné en 1993. La mutation des allèles défectueux est une expansion d'un triplet CAG (région
5' du gène) qui modifie la protéine codée par ce gène : l'huntingtine. Lorsque la répétition
comporte moins de 30 motifs, la protéine est fonctionnelle, entre 30 et 39 répétitions, le
phénotype est variable et plus de 39 répétitions du motif altèrent la protéine. Puisque cette
maladie est dominante sur le phénotype sain il semble que l'allongement de la protéine doit lui
conférer des propriétés différentes de la protéine normale (nouvelle activité ou expression
modifiée). Plus le nombre de répétitions est élevé plus la maladie se déclare tôt. Comme le
nombre de répétitions peut varier des parents aux enfants on ne peut prévoir le degré de
gravité chez les descendants.
La détection de l'allèle muté se fait par PCR en amplifiant l'ADN total du sujet avec deux
amorces oligonucléotidiques uniques qui encadrent les motifs répétés. La taille du fragment
amplifié est déterminée par électrophorèse sur gel.
2. Détection indirecte
Dans d'autres cas si l'on connaît le gène dont une mutation est responsable du phénotype
[malade], on n'a pas identifié la mutation. Il est donc impossible de la détecter. Dans de tels
cas on va chercher un marqueur qui permettra d'identifier le chromosome (la portion de
chromosome proche du gène) qui porte l'allèle muté pour déterminer si c'est lui qui a été
transmis au descendant. Pour pouvoir connaître l'état de cette portion du génome des parents
il faut avoir examiné un malade de la même famille (enfant de la même fratrie, ascendant ,
cousin, ...)
Phénotype dominant
L'aniridie est une malformation de l'oeil qui est dépourvu d'iris. Cette malformation
s'accompagne de perte sévère d'acuité visuelle. C'est une maladie rare (1 naissance sur 100
000. La figure 8-6 permet de déduire que ce phénotype est dominant sur le phénotype normal
et que le gène concerné est autosomique.

Figure 8- 7.Pedigree d'une famille présentant plusieurs cas d'aniridie.
De nombreuses mutations sont la cause de ce défaut. Elles affectent un gène important dans le
programme de développement des organismes multicellulaires (Pax6 localisé sur le bras court
du chromosome 11, bande 13). Il ne peut donc être question de repérer chaque mutation
individuellement. La stratégie de diagnostic prénatal suivie dans ce cas a été de trouver un
marqueur polymorphe (un RFLP qui présente 2 formes) suffisamment proche de Pax6 pour
que l'on puisse raisonnablement négliger la faible probabilité de Crossing Over entre le site
RFLP et le site de la mutation. Puisque les deux sites n'ont qu'une probabilité très faible de
recombiner, tous les gamètes fournis seront de type parental. On détermine quel est le
génotype du parent atteint pour ce marqueur et le gène Pax6. L'examen du génotype de
l'embryon permet de dire quel chromosome 11 lui a été transmis.
Figure 8- 7. Région du chromosome 11 contenant le gêne Pax6 et le site polymorphe
AvaI.
Dans la famille de la figure 8-8 un second enfant est attendu. Sera-t-il atteint d'aniridie ?
Chaque personne de la famille a été testée pour l'état du site AvaI . Pour cela , on a prélevé un
peu de sang. Des globules blancs on a extrait l'ADN total. Celui-ci a subi une digestion
complète par AvaI. Tous les fragments obtenus ont été classées par ordre de taille avec une
électrophorèse sur gel d'agarose. Les fragments d'ADN ainsi rangés sont transférés sur
membrane de nylon (Southern Blot) afin que l'on puisse les hybrider avec la sonde unique
marquée S1(de l'ordre de 300bp). Parmi le million (valeur approximative) de bandes résultant
de la digestion par AvaI, seules celles qui ont une séquence identique à S1 vont retenir la
sonde par hybridation. Sur une autoradiographie elles seules seront détectées. On saura donc
si le sujet testé présente une bande de 6,5kbp (le site polymorphe est absent sur les deux
chromosomes homologues) ; une bande de 4kbp (le site polymorphe est présent sur les deux
chromosomes homologues) ou les deux bandes (le site polymorphe est absent sur l'un et
présent sur l'autre)

Figure 8- 8. Diagnostic prénatal de l'aniridie. A: pedigree de la famille, B : analyse du
site polymorphe très proche du gène Pax6, C : reconstitution des deux chromosomes 11
homologues paternel et maternel reçus par II-1, D : reconstitution des deux
chromosomes 11 portés par la mère.
II-1 qui porte un site AvaI présent et un absent (Av+/Av-) ne peut avoir reçu Av+ que de sa
mère. C'est également elle qui lui a fourni l'allèle muté Pax6-. On peut donc écrire son
génotype .
La mère a pour phénotype [aniridie , bandes 4 et 6,5]. Elle est donc hétérozygote pour le gène
pax6 (pax6-/pax6+) et pour le site polymorphe (AvaI-/AvaI+). En l'absence d'informations
supplémentaires il y a deux façons possibles d'écrire son génotype, deux phases pour le site
polymorphe et le gène : .
Sachant ce que son premier enfant a reçu d'elle (AvaI+, pax6-) permet d'éliminer la première
possibilité puisque site polymorphe et gènes sont si proches qu'un CO entre eux est très
improbable.
Un tel test nécessite que le parent porteur de la maladie soit hétérozygote également pour le
site polymorphe examiné, ce qui n'est pas toujours le cas (n'oubliez pas que ce site
polymorphe n'a rien à voir avec la maladie, il est juste à côté du gène concerné)
1
/
5
100%