enzymologie. - BDE Sciences
publicité

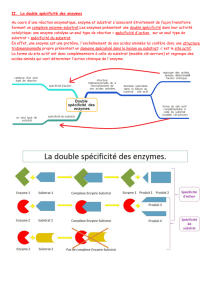
ENZYMOLOGIE. Q U ’ EST - CE QU ’ UNE ENZYME ? Les enzymes représentent l’ensemble de protéines fonctionnelles les plus importantes. Enzyme = catalyseur. Leur rôle est d’accélérer une réaction. Pour donner un ordre de grandeur, dans une cellule, à chaque instant, il y a environ 10 000 réactions en cours. Les enzymes sont spécifiques. C'est-à-dire, qu’elles vont catalyser une seule et unique réaction, contrairement aux catalyseurs artificiels. Elles sont aussi très efficaces. Elles accélèrent les réactions d’un facteur 103 à 106 !!! Leur seule limite : elles ne fonctionnent que dans un domaine restreint de température, de pression, de pH. Parce que 99,99999% des enzymes sont des protéines. S PECIFICITE ENZYMATIQ UE . Les enzymes peuvent être caractérisées par une spécificité stricte. EXEMPLE DES PROTEASES DIGESTIVES. Dans notre alimentation, on ingère une grande quantité de protéines sous forme repliées, tridimensionnelle, etc. Une fois dans l’estomac, les protéines vont être digérées, dénaturées par l’acide. Une fois dénaturées, dans l’intestin, des enzymes digestives vont protéolyser pour transformer les protéines en acides aminés. C’es enzymes sont soit des endopeptidases : elles coupent au beau milieu de la chaine peptidique. Soit ce sont des exopeptidases, et elles grignotent un à un les acides aminés d’une extrémité à l’autre. Le fait d’avoir des endopeptidases ET des exopeptidases accélère encore plus le processus : l’endopeptidase crée des extrémités où l’exopeptidase pourra agir. On appelle ça, le travail parallèle. EXEMPLE DE LA TRYPSINE. Elle va venir catalyser l’hydrolyse d’un polypeptide. Mais seulement après un acide aminé à chaine latérale basique (lysine, asparagine, histidine). Elle n’hydrolyse que les liaisons peptidiques. Et pas n’importe quelle liaison peptidique : qu’après (pas avant) un acide aminé chargé +. En plus, elle ne peut se charger que des peptides de série L (pas de série D). 1 EXEMPLE DE LA CHYMOTRYPSINE. Elle agit d’une façon semblable, mais la chymotrypsine ne coupe qu’après un des 3 acides aminés aromatiques (phénylalanine, tyrosine, tryptophane). Et elle ne fonctionne que sur la série L. EXEMPLE DES EXOPEPTIDASE. Elles débitent les protéines en acides aminés, à partir d’une des deux extrémités de la chaine polypeptidique. Si elles reconnaissent l’extrémité COO-, ce sont des carboxypeptidases. Dans l’autre cas, on les appelle aminopeptidases. Dans ce deux cas, on ne reconnaît plus la chaine latérale, mais l’extrémité. EXEMPLE DES ENZYMES QUI AGISSENT SUR L’ADN. Comme l’ADN Polymérase. Elle sait créer des liaisons phospho-diester (qui se fait entre le carbone 3’ d’un désoxyribose et le 5’ du suivant). Les endonucléases de restriction coupent une liaison phospho-diesters qui coupent des palindromes (voir cours de génie génétique). E FFICACITE ENZYMATIQU E . Il faut bien garder à l’esprit que les enzymes ne font qu’augmenter la vitesse d’une réaction. NOTION DE VITESSE DE REACTION. On travaille sur un ensemble de molécules, pas que sur une seule molécule. On ne sait pas estimer en combien de temps une molécule de A donne une molécule de B. On ne s’intéresse qu’à de grand ensemble de protéines. POUR UNE REACTION DE TYPE AB. Vitesse = nombre de molécules de A transformées en B par unité de temps. 𝑣= 𝑑𝐴 𝑑𝐵 = = 𝑘[𝐴] 𝑑𝑡 𝑑𝑡 2 POUR UNE REACTION DE TYPE A + B C 𝑣 = 𝑘 𝐴 . [𝐵] DANS LE CAS DES EQUILIBRES DE TYPE : A + B C + D On a 𝑣1 = 𝑘1 𝐴 . [𝐵] et 𝑣−1 = 𝑘−1 𝐶 . [𝐷] et 𝑣1 = −𝑣−1 et 𝑘1 𝐶 . [𝐷] = = 𝑘𝑒𝑞 𝑘−1 𝐴 . [𝐵] La vitesse de réaction est corrélée par ce qu’on appelle l’énergie d’activation. C’est l’énergie nécessaire à la réaction pour se produire. En fait les molécules bougent. Elles ont de l’énergie cinétique. Celles qui vont réagir sont celles qui ont une énergie cinétique supérieure ou égale à l’énergie d’activation. Pour ce, on peut augmenter la température. Ce qui agite encore plus les molécules. Ce qui facilite l’atteinte de l’énergie d’activation. Même si le ΔG est favorable, les molécules passent par un intermédiaire à haut niveau d’énergie, avant d’atteindre un niveau d’énergie plus bas (ΔG<0). M ECANISME DE CATALYSE ENZYMATIQUE . L A RECONNAISSANCE ENZYMATIQUE . S’il y a spécificité, il doit y avoir un mécanisme de reconnaissance moléculaire. Il faut que les liaisons entre l’enzyme et le substrat soient facilement réversible. On ne met donc pas en place des liaisons covalentes, mais plutôt des liaisons hydrogènes, Van Der Waals (ou liaisons hydrophobes), liaisons ioniques (mais c’est très rare car ces liaisons restent « trop fortes » on trouvera ces dernière plus souvent à l’intérieur des structures protéiques). Nécessairement, une partie de l’enzyme va interagir avec les intermédiaires réactionnels. Et ce, probablement par l’intermédiaire des ses groupement OH, NH, COOH… de ses acides aminés. L’enzyme va « prêter » ses groupements fonctionnels, et les récupérer à la fin de la réaction. Parfois les enzymes ont besoin de quelques choses d’un peu plus chargé, un peu plus réactif, donc elles empruntent des métaux (genre Fe2+) pour aider à la catalyse. Ce qui est trop énorme, c’est qu’avec une vingtaine d’acides aminés, on est capable de former des milliers d’enzymes qui catalysent des milliers de réactions différentes. On appellera « site actif » la partie de l’enzyme où la reconnaissance se fait et où la catalyse a lieu. 3 EXEMPLE DE L ’ACETYLCHOLINESTERASE (ACHE). L’acétylcholine est un neurotransmetteur qui agit à deux niveaux. Au niveau du système nerveux central et du système nerveux périphérique. Dans le système nerveux périphérique c’est le neurotransmetteur de la contraction musculaire squelettique. En fait, on a des extrémités axoniques qui sont à la jonction de la plaque motrice. Lorsque le potentiel d’action arrive, la dépolarisation au niveau terminal entraine le relargage d’acétylcholine. Puis, l’acétylcholine vient se fixer au récepteur de l’acétylcholine sur le muscle. Ce récepteur a une fonction bien particulière : c’est un récepteur canal. Lorsque l’acétylcholine se fixe à ce récepteur, il change de conformation et laisse entrer des ions. La cellule étant chargée négativement, lorsqu’on laisse entrer de Na+ et du Ca++, se dépolarise. D’autres canaux, sensibles à cette dépolarisation, libèrent beaucoup de Ca++, ce qui entraine la contraction des muscles. Ces récepteurs sont inhibés par la nicotine. D’ailleurs, la nicotine agit au niveau du système nerveux central par inhibition des récepteurs à l’acétylcholine. Il faut bien un mécanisme qui arrête le mécanisme, sinon, il y aurait un tétanos. Donc, après la contraction, il faut que la concentration en acétylcholine diminue vite et beaucoup. Dans les jonctions neuromusculaires, on trouve beaucoup d’une enzyme : acétylcholinestérase. L’acétylcholine est relativement stable et il faut vite fait la dégrader. L’acétylcholine est un acétyle et une choline liés par une liaison ester. Cette liaison est hydrolysable. Mais la molécule est trop stable pour que l’eau, présente dans le milieu, puisse lyser la liaison facilement. Le O avec la double liaison tourne autour du carbone de la liaison ester et empêche l’eau d’y accéder. L’ammonium quaternaire hydrophobe (à cause des 3 méthyles qui lui sont accrochés) est facilement reconnaissable. Donc si on devait créer l’enzyme capable de catalyser la lyse de l’acétylcholine. Dans un premier temps, pour se fixer à l’ammonium quaternaire, on pourrait penser à utiliser des acides aminés à chaines latérale acide (aspartate ou glutamate). Mais on va éviter car la liaison serait trop forte et on ne pourrait plus séparer l’enzyme du substrat. Alors on va avoir recours à des acides aminés à chaine latérale aromatique. Et dans l’acétylcholinestérase on a en effet une cavité avec des aromatique. Ça c’est pour le site de reconnaissance. Pour le site catalytique, il y aura une sérine qui viendra à proximité du carbone de la liaison ester. Cet acide aminé est soutenu par une histidine et un aspartate, disposés à sa proximité, qui ont tendant à capter un hydrogène de la sérine. Le doublet libre, du O de la sérine va attaquer le carbone de la liaison ester, qui va rompre l’une des deux liaisons de la double liaison avec le O de la choline. A ce moment on a une liaison ester entre l’acétyle et la sérine, on a un intermédiaire covalent : l’acétyle-enzyme. La structure est particulièrement rigide, et l’eau agit maintenant beaucoup beaucoup plus facilement. 4 Et on se retrouve avec notre choline d’une part, notre acétate d’autre part, et l’enzyme comme au commencement. L’étape limitante de la réaction est la rencontre de l’eau avec le carbone de la liaison ester. Le fait de faire intervenir l’enzyme coute moins cher en énergie. L’enzyme abaisse en fait l’énergie ‘activation nécessaire à la lyse. Les inhibiteurs de l’acétylcholinestérase. Ils appartiennent à la famille des organophosphorés, où on retrouve les insecticides et gaz de combat. Ces inhibiteur ont un phényle substitué qui va se fixer au site de reconnaissance de l’enzyme et les O du phosphate vont dans le site catalytique. Ensuite, la partie organique avec le phénol se détache et il reste, dans le site catalytique le phosphate : intermédiaire phosphate-enzyme. C’est un inhibiteur covalent. Mais pour éviter qu’on ne tue tout l’immeuble pour tuer des insectes, on joue sur les substituants. On utilisera des substituants qui n’attaqueront que les acétylcholinestérases des insectes. M ESURE DE L ’ ACTIVITE ENZYMATIQUE . M ECANISMES DE FONCTIO NNEMENT Pour mesurer l’activité, on dose les concentrations en substrat et en produit. DOSAGE EN CONTINU . On a recours à des dosages en continu dans les cas où la technique de dosage n’a pas d’influence sur la réaction. Par exemple les lectures de D.O. Le produit de la phosphatase alcaline par exemple a une couleur jaune, tandis que le substrat est incolore. 5 DOSAGE EN DISCONTINU. On prend comme exemple la mesure de l’ADN Pol. Par l’intermédiaire de DxTPs radioactifs. Mais comme la quantité de radioactivité du tube est constante, on ne peut pas avoir recours à un dosage en continu. Donc on prélève à intervalle régulier un peu de notre solution et on l’étudie. MESURE DES VITESSES. On fait des mesures. On reporte les données ponctuelles sur un graphe, et on ne relie pas les points par des segments de droites, mais on doit obtenir une courbe continue et arrondie. Pour traiter les données on utilise des outils spécifiques à l’enzymologie, qui nous viennent des mathématiques. Voilà ce qui se passe : 𝐸+𝑆 𝑘1 𝑘 −1 𝐸𝑆 𝑘2 𝐸+𝑃 Ce qui est important est de connaitre nos constantes k. Dans la courbe, on remarque que la pente (qui est proportionnelle à k) diminue avec le temps jusqu’à devenir nul, logique : au bout d’un certains temps, on a consommé tout le substrat. A T0 on relève la première tangente. On remarque que pendant les premiers instant la courbe se superpose à la tangente : la vitesse initiale reste constante un instant. Ceci est dû au fait qu’on met peu d’enzyme et beaucoup de substrat, aussi, vu les proportions, on peut considérer que pendant quelques instants [S] ne change pas. On a : 𝑣𝑖 = 𝑑 𝑃 = 𝑘2 [𝐸𝑆] qui est constant dans l’état initial (appelé état stationnaire). 𝑑𝑡 Ce qui implique (vu que K2 est une constante) que [ES] est constant. C’est à dire qu’on a autant de complexe ES généré que de complexe ES défaits. On a : 𝑘1 𝐸 𝑆 = 𝑘−1 𝐸𝑆 + 𝑘2 𝐸𝑆 = 𝑘−1 + 𝑘2 . 𝐸𝑆 . 𝑘 +𝑘 On trouve : 𝐸 . 𝑆 [𝐸𝑆] = −1 2 𝑘 = 𝐾𝑚 = 𝑐𝑜𝑛𝑠𝑡𝑎𝑛𝑡𝑒 𝑑𝑒 𝑀𝑖𝑐ℎ𝑎𝑒𝑙𝑖𝑠 1 𝑣 = 𝑘2 𝐸𝑆 = 𝑘2 𝐸𝑆 = 𝐾𝑚 𝑆 +1 𝐸𝑆 = 𝐸0 𝐸0 𝐸𝑆 𝐾𝑚 +[𝑆] [𝑆] 𝐸𝑆 𝑒𝑡 𝐸0 = 𝐸 + 𝐸𝑆 𝑒𝑡 𝐾𝑚 = 𝐸 . 𝑆 [𝐸𝑆] 𝑑𝑜𝑛𝑐 ∶ 𝑒𝑡 𝑜𝑛 𝑟𝑒𝑝𝑜𝑟𝑡𝑒 ç𝑎 𝑑𝑎𝑛𝑠 𝑙 ′ é𝑞𝑢𝑎𝑡𝑖𝑜𝑛 𝑑𝑒 𝑣𝑖𝑡𝑒𝑠𝑠𝑒: 𝑣 = 𝐸0 = 𝐾𝑚 [𝑆] 𝐸𝑆 + 𝐾2 𝐸0 𝑆 𝐾𝑚 +[𝑆] Si on fait varier la concentration en substrat, on obtient différentes valeurs de v i. On dessine la courbe de la vitesse en fonction de la concentration en substrat. La limite quand [S] tends vers l’infini est un plateau Vmax=𝑘2 [𝐸0 ]. Grâce à cela on peut trouver la valeur de K M. On prend la concentration correspondant à l’ordonnée 1/2Vmax. Parfois la courbe ne permet pas de visualiser V max. Dans ces cas, on linéarise la courbe en 1 𝐾𝑚 1 1 prenant son inverse : = × + 𝑣 𝑉𝑚 𝑆 𝑉𝑚𝑎𝑥 6 On obtient une droite de type ax+b. On voit qu’on croise l’axe des ordonnées à la valeur 1/Vmax, et on a une pente de Km/Vmax, et on croise l’axe des abscisses à la valeur -1/Km. I NHIBITION ENZYMATIQUE . I NHIBITEUR COMPETITIFS . Dans ce cas, l’inhibiteur se lie à l’enzyme à la place du substrat. On définit une constante Ki, qui est d’autant plus petite que l’inhibiteur est efficace. 𝐸 𝐼 On a : 𝐸0 = 𝐸 + 𝐸𝐼 + 𝐸𝑆 et 𝐾𝑖 = Donc 𝐸0 = 𝐾𝑚 𝑆 1+ 𝐼 𝐾𝑚 donc 𝐸0 = 𝐸 . (1 + 𝐸𝐼 𝐼 ) 𝐾𝑖 𝐾𝑚 ×(1+ 𝐸𝑆 + 𝐸𝑆 = 𝑠 +1 𝐸𝑆 = 𝐼 𝐾𝑖 ) et 𝐸 = 𝐼 +[𝑆] 𝐾𝑖 𝐾𝑚 × 1+ 𝑠 𝐾𝑚 [𝑆] 𝐸𝑆 . 𝐸𝑆 . Si on reporte dans l’équation de vitesse : 𝑣= 𝑘2 𝐸0 𝐸𝑆 On a 𝐾𝑚′ = 𝐾𝑚 (1 + l’enzyme. 𝐼 𝐾𝑖 = 𝐼 𝐾𝑚 . 1 + + [𝑆] 𝑘𝑖 × [𝐸𝑆] [𝑆] 𝐾2 𝐸0 𝑆 𝐾𝑚 . 1 + 𝐼 + [𝑆] 𝐾𝑖 ). Et on cherche le KI : la valeur de l’affinité de l’inhibiteur pour Si on développe Km’, on a 𝐾𝑚′ = 𝐾𝑚 + [𝐼] 𝐾𝑚 𝐾𝐼 . Qui est de forme ax+b. On a donc une représentation graphique sous forme de droite de coefficient directeur Km/KI. Si pour une concentration en substrat donnée, on divise le K m par le coefficient directeur de la droite, on obtient le KI I NHIBITEUR NON - COMPETITIVE . Le substrat se lie à l’enzyme dans un site différent de l’inhibiteur. L’inhibiteur, lorsqu’il se fixe, modifie la conformation de l’enzyme. Résultat : le complexe ESI se forme mais est totalement inefficace. Sur ES on peut fixer I et sur EI on peut fixer S. La constante K 1 de liaison de E+S en ES est égale à la constante de liaison de EI+S et EIS. Km n’est donc pas modifié. C’est la vitesse maximale qui se trouve changée. On a : 𝑉 ′ 𝑆 𝑉 ′ 𝑣 = 𝑚𝑎𝑥 et 𝑉𝑚𝑎𝑥 = 𝑚𝑎𝑥 𝐼 𝐾𝑚 + 𝑆 1+ 𝐾𝐼 On a : 𝑣 = 𝐾2 𝐸𝑆 = 𝐾2 𝐾𝐼 = 𝐸 𝐼 𝐸𝐼 = 𝐸𝑆 𝐼 𝐸𝑆𝐼 . On a 𝐸0 𝐸0 et [𝐸𝑆] 𝐸𝐼 = 𝐸 × 𝐼 𝐾𝐼 et [𝐸0 ] = 𝐸 + 𝐸𝐼 + 𝐸𝑆 + [𝐸𝑆𝐼] 𝐸𝑆𝐼 = 𝐸𝑆 × 𝐼 𝐾𝐼 et 𝐾𝑚 = 𝐸 𝑆 𝐸𝑆 et . 7 On remplace : 𝐸0 = 𝐸 + 𝐸𝐼 + 𝐸𝑆𝐼 + 𝐸𝑆 𝐸 𝐼 𝐸𝑆 𝐼 + + [𝐸𝑆] 𝐾𝑖 𝐾𝑖 𝐸0 = 𝐸 + 𝐸0 = 𝐾𝑚 𝐸𝑆 𝐾𝑚 𝐸𝑆 𝐼 𝐸𝑆 𝐼 + + + 𝐸𝑆 𝑆 𝑆 𝐾𝑖 𝐾𝑖 𝐸0 = 𝐸𝑆 ( 𝐾𝑚 𝐾𝑚 𝐼 𝐼 + + + 1) 𝑆 𝑆 . 𝐾𝑖 𝐾𝑖 𝐾𝑚. 𝐾𝑖 + 𝐾𝑚 𝐼 + 𝑆 𝐼 + 𝑆 𝐾𝑖 𝐸0 = 𝐸𝑆 ( ) 𝑆 𝐾𝑖 𝐾𝑚 𝐼 + 𝐾𝑖 + 𝑆 𝑆 𝐾𝑖 𝐸0 = 𝐸𝑆 ∗ 𝐾𝑚 + 𝑆 𝐼 + 𝐾𝑖 𝑆 𝐾𝑖 𝐸0 = 𝐸𝑆 𝑉= 𝐼 + 𝐾𝑖 𝑘2 𝐸0 𝐸𝑆 𝑘2 𝐸0 𝑆 →𝑉= 𝐾𝑚 + 𝑆 𝐼 + 𝐾𝑖 𝐼 + 𝐾𝑖 𝐸𝑆 ∗ (𝐾𝑚 + 𝑆 ) 𝐾𝑖 𝑆 𝐾𝑖 𝑉= 𝑘2 𝐸0 𝑆 𝑉𝑚𝑎𝑥 𝑆 →𝑉= ∗ [𝐼] 𝐼 𝐾𝑚 + [𝑆] 1+ ∗ (𝐾𝑚 + 𝑆 ) 1+ 𝐾𝑖 𝐾𝑖 Avec un inhibiteur non-compétitif, on modifie la valeur de Vmax , mais pas le Km. Vmax prend la valeur suivante : 𝑉𝑚𝑎𝑥′ = 𝑉𝑚𝑎𝑥 𝐼 1+ 𝐾𝑖 I NHIBITION COVALENTE . Formation d’un complexe ES d’une part et de EI d’autre part (EI non fonctionnel). La vitesse de disparition de E : − 𝑑 𝐸 𝑐𝑠𝑡𝑒 A t=0 on a ln 𝐸 = −𝑘 𝐼 𝑡 + ln 𝐸0 𝑑𝑡 =𝑘 𝐸 𝐼 ↔ 𝐸 𝐸0 = 𝑒 −𝑘 ↔ 𝑑 𝐸 𝐸 = −𝑘 𝐼 𝑑𝑡 ↔ ln 𝐸 = −𝑘 𝐼 𝑡 + 𝐼𝑡 8 Pour mesurer une réaction d’inhibition covalente, on part de ce qu’on connait : 𝑣= 𝑘2 𝐸0 𝑆 𝐾𝑚 + 𝑆 La vitesse est proportionnelle à E0. A intervalles réguliers on prélève des échantillons de notre mélange réactionnel et on ajoute du substrat, et on analyse la vitesse pour savoir il nous reste combien d’enzyme dans notre prélèvement. Cela n’est utile que si on arrête la réaction E+I=EI, à chaque prélèvement. EXEMPLE D’INHIBITEUR COVALENT . Il s’agit de petites molécules réactives avec certaines chaînes latérales. Par exemple, les sérines et les cystéines sont très réactives. On va étudier un inhibiteur à cystéine : l’iodoacétamide : I-CH2-CONH2. Cette molécule va perdre son iode, et se lier au souffre d’une cystéine portée par une enzyme à cystéine. Ces inhibiteurs manquent de spécificité : on va si lier à toutes les cystéines. Ce qui est pratique en laboratoire c’est lorsqu’on veut inhiber toues les protéases présente. Ça inhibe toutes les enzymes. Ça peut aussi être utile pour analyser le fonctionnement d’une enzyme : on met une enzyme, son substrat et de l’iodoacétamide. Si rien ne se passe, on sait que l’enzyme fait intervenir une cystéine. On va mettre beaucoup de substrat pour savoir si l’acide amine concerné se trouve sur le site de reconnaissance ou sur le site actif. E FFETS DE L ’ ENVIRONNEMENT SUR LES ENZYMES . On parle de l’environnement physico-chimique. Il y a deux paramètres importants : le pH et la température. Ces paramètres vont influencer les chaînes ionisable des acides aminés (au niveau conformationnel et sur les sites de reconnaissance et catalytique). Ces paramètres ont des conséquences sur la structure et sur les mécanismes réactionnels. L ES REPLIEMENT DES PR OTEINES . En temps normal, on a une structure primaire et la molécule va se replier. En étudiant les mécanismes de dénaturation de la protéine, on peut considérer qu’il existe un équilibre qui tend vers la formation de la forme bien repliée de la protéine, avec un ΔG peu négatif (-5kcal.mol-1). On considère ∆𝐺 = −𝑅𝑇 ln 𝐾𝑒𝑞 avec 𝐾𝑒𝑞 = On a 𝑁 𝐷 =𝑒 −∆𝐺 𝑅𝑇 𝑓𝑜𝑟𝑚𝑒 𝑛𝑎𝑡𝑖𝑣𝑒 𝑟𝑒𝑝𝑙𝑖 é𝑒 𝑓𝑜𝑟𝑚𝑒 𝑑é𝑛𝑎𝑡𝑢𝑟 é𝑒 . Le Keq est de l’ordre de grandeur d’une liaison hydrogène. 9 P H ET STRUCTURES DES EN ZYMES . Le pH influe sur les charges des groupements ionisables. Ce qui va modifier les liaisons hydrogènes et ioniques (ou ponts salins). Lorsqu’on est au pH=pKa, on a 50% de la forme protonnée et 50% de la forme déprotonée. A chaque fois qu’on s’éloigne d’une unité de pH (vers le bas pour les acides aminés à chaines latérales basiques et vers le haut pour ceux à chaines latérales acides), on augmente jusqu’à 90% la proportion d’enzyme active, si on change de 2 unités, on arrive à 99 %. Si on trace la courbe de V max de l’enzyme (en %) en fonction du pH on va avoir une courbe en forme de cloche. 100 90 80 70 60 50 40 30 20 10 0 La zone du milieu, où l’enzyme est efficace à 90% est appelée « zone de sécurité ». I NFLUENCE DE L ’ ENVIRONNEMENT DES AC IDES AMINES SUR LES VALEUR DE P K A . Lorsque deux acides aminés à chaine latérale acide, sont proches, quand on les met à un pH de 7, on se dit qu’ils seront tous les deux déprotonnés. Mais deux charges négatives si proches risquent se repousser, ce qui serait énergétiquement défavorable. La nature remédie à cela en laissant les deux chaines latérales avec leurs hydrogènes. Ce qui est favorable puisqu’en plus, on des liaisons hydrogènes pour stabiliser le tout. Si on a dans une enzyme, une poche hydrophobe contenant un groupement COOH, on va assister à l’augmentation du pKa, pour ne pas perdre le H du COOH. P H ET FONCTIONNEMENT EN ZYMATIQUE . 10 On s’intéresse à ce qui va se passer dans le site actif, composé du site de reconnaissance et du site catalytique. ACIDE AMINE IONISABLE DANS LE SITE DE RECONNAISSANCE. On considère la présence d’un COO- dans le site. Il est fait pour reconnaitre les molécules chargées positivement. Selon le pH, ce COO - peut devenir COOH, comme si l’ion H+ était un inhibiteur compétitif ! ACIDE AMINE IONISABLE DANS LE SITE CATALYTIQUE. On considère une protéine qui possède au niveau de site catalytique deux acides aminés ionisables. D’une part, un COO- et une cystéine (avec un groupement thiol SH), de pKas respectifs de 4,5 et 8,5. Ainsi, à pH 4,5 on a 50% d’activité pour le COO - et à pH 8,5 on a 50% d’activité pour la cystéine. A pH 5,5 on a 90% d’activité pour le COO - et à pH 7,5 on a 90% d’activité pour la cystéine. Ainsi on voit qu’à pH 4,5 et à pH 8,5 on 50% d’activité pour le site catalytique (si on le réduit à ces deux seuls acides aminés). Et entre les deux, on a la zone de sécurité. T EMPERATURE ET FONCTIONNEMENT ENZYMATIQUE . L’augmentation de la température est censée augmenter l’activité enzymatique (augmentation de l’agitation moléculaire). Mais si on chauffe trop, on risque de trop agiter les atomes d’une enzyme, ce qui va la dénaturer. Pour empêcher la dénaturation par la chaleur, on peut stabiliser l’enzyme en la mettant tout simplement en présence de son substrat. Lorsqu’elle est liée à son substrat par des liaisons hydrogènes, ça la stabilise. On peut aussi stabiliser une enzyme à l’aide de sels divalents. I NTERACTIONS LIGAND - RECEPTEUR . Les organes communiquent entre eux par le biais de signaux (petites molécules). Ces signaux ont peuvent avoir pour cible des récepteurs membranaires dans 45% des cas (dont 2% de récepteurs de la membrane nucléaire), ou des enzymes dans 28% des cas. Ces récepteurs sont des « antennes » cellulaires qui, lorsqu’ils reçoivent un signal, déclenchent des réactions à l’intérieur de la cellule. Il existe des ligands liposolubles (hydrophobes) qui peuvent diffuser librement dans les membranes. Ils se lient en dimère avec les récepteurs. Ils arrivent jusqu’au noyau et induisent généralement la transcription de gène. 11 Les ligands hydrosolubles (polaires) ne peuvent pas passer simplement à travers la membrane. Ils interagissent donc avec un site polaire d’un récepteur membranaire, qui peut être de différents types : - les récepteurs enzymatiques : ils possèdent un site intracellulaire qui possède une activité enzymatique. - les récepteurs couplés à une enzyme : la reconnaissance du signal par le récepteur entraine l’activation d’une molécule intracellulaire qui a une activité enzymatique. - les récepteurs couplés à une protéine G : la reconnaissance du signal par le récepteur entraine l’activation d’une protéine G. - les récepteurs canaux : la reconnaissance du signal par le récepteur entraine l’activation d’un canal qui laisse entrer des ions, ce qui a tendance à faire varier le potentiel membranaire. On a, en guise de première étape : 𝑅 + 𝐿 ⇋ 𝑅𝐿 La liaison entre ligand et récepteur se fait par le biais de liaisons Hydrogènes, ou d’interaction de Van Der Waals. On n’observe pas de liaison covalente. La deuxième étape est l’étape qui est induite par le signal, à l’intérieur de la cellule. Comme avec les réactions enzyme-substrat, on a une constante de Michaelis, généralement comprise entre 10-8 M et 10-12 M. 𝑘𝑑𝑥 𝑘𝑎𝑥 On veut connaitre le nombre de sites récepteurs par ligand et leur affinité : 𝐾𝑑𝑥 = 𝐾𝑑 = 𝑅 𝐿∗ 𝑅𝐿∗ O UTILS TECHNIQUE : LIGAND/RECEPTEUR. Pour suivre l’évolution de l’établissement de liaison, on utilise un ligand radioactif pur. Il faut veiller à ce que la radioactivité n’influe pas sur l’activité du récepteur. SOURCE DES RECEPTEURS. L’étude de la source des récepteurs est plus rare. On l’étudie souvent dans l’analyse de cellule, et moins souvent dans l’analyse d’enzymes. On utilisera soit la membrane en tout ou en partie, voire la cellule entière. 12 LA DIALYSE A L ’EQUILIBRE. On met des récepteurs dans le compartiment A et des ligands dans le compartiment B. Les deux compartiments sont séparés par une membrane semi-perméable. Les récepteurs ne peuvent pas traverser la membrane, tandis que les ligands le peuvent. Par osmose, les ligands vont passer de B à A. Les premiers ligands qui arrivent dans A vont se lier aux récepteurs, puis d’autres vont suivre, pour équilibrer la concentration en ligand libre dans les deux compartiments. Ensuite, on mesure la radioactivité et on fait la différence entre les deux compartiments pour connaitre la concentration en récepteur. Exemple : au final on a 10010 cpm en A et 10000 cpm en B on sait que les 10 cpm proviennent des complexes RL*. 13 LA CENTRIFUGATION. On met dans un tube des récepteurs et des ligands. On attend. On centrifuge et on se retrouve avec un culot et un surnageant. Le surnageant ne contient que des ligands. Le culot contient des complexes RL et des ligands libres aussi. Pour isoler les RL des ligands dans le culot, on pourrait essayer de laver le culot, mais ça risque de dissocier les RL en R + L. L’ULTRAFILTRATION. On a un filtre avec des mailles adaptées à ce qu’on veut retenir. On veut laisser passer le ligand libre et retenir les RL. Mais en général, on retient au passage les membranes, les fragments de cellules, etc. On peut aussi faire une ultrafiltration à haute vitesse (on ajoute une aspiration pour optimiser le processus). Dans ce cas on a le ligand qui passe et les complexes RL qui restent bloquées. L’avantage de l’ultrafiltration c’est qu’on n’a pas de dissociation de RL. Problème par rapport aux liaisons spécifiques une partie du ligand va se fixer dans la membrane sans aller au récepteur ce qui fausse les résultats. Pour remédier à cela, on effectue deux expériences : -on met des morceaux de membranes contenant des récepteurs dans un tube, et on ajoute du ligand marqué radioactivement. Les ligands se lient de manière spécifique aux récepteurs. Mais certains ligands se lient de manière non-spécifique à la membrane. On évalue la radioactivité et on connait, dans un premier temps, la quantité de RL* et de ligand lié à la membrane. -ensuite, on ajoute à tout ça des ligands non-marqués en large excès. Ces ligands nonmarqués vont se lier, mais vu qu’ils sont en large excès, ils vont prendre la place de ligands marqués liés de façon spécifique (aux récepteurs). On centrifuge, et on prend notre culot qui contient des fragments de membrane avec des récepteurs liés à des ligands non-marqués et les fragments marqués sur les membranes liés de façon non-spécifique. On connait maintenant la quantité de ligands liés de façon non-spécifique. On fait la différence et hop-là ! On a notre quantité de RL* !!! Le problème reste de savoir combien de temps on doit laisser les ligands se lier. Si on fait des prélèvements à intervalles réguliers on a de plus en plus de RL*, jusqu’à atteindre un plateau, un équilibre. Et ce n’est qu’à ce moment là, qu’on ajoute notre excès de ligands non-radioactifs. On peut calculer une constante de vitesse d’association (K a) et de dissociation (Kd). 𝐾= 𝑘𝑑 𝑘𝑎 𝑅𝐿 = 𝑅 0 . 𝐿 𝐾𝑑 + 𝐿 [RL] sera au max quand tous les récepteurs seront liés à un ligand. On a [RL]max=R0 et R0/2 = Kd. 14 R EPRESENTATION DE S CATCHARD . 𝑅 𝐿 𝑒𝑡 𝑅0 = 𝑅 + 𝑅𝐿 [𝑅𝐿] 𝑅0 𝑅𝐿 𝑅𝐿 𝑅𝐿 1 𝑅0 ↔ − = ↔ = × 𝑅𝐿 + 𝐾𝑑 𝐾𝑑 𝐿 𝐿 𝐾𝑑 𝐾𝑑 𝐾𝑑 = 𝑅0 − 𝑅𝐿 𝐿 = 𝐾𝑑 𝑅𝐿 Linéarisation de Scatchard [RL]/[L] L’intersection avec l’axe des abscisses = R0 𝑅𝑡 L’intersection avec l’axe des ordonnées = La pente = -1/Kd 𝑘𝑑 On peut, à l’aide de ce genre de représentations, comparer des récepteurs avec des ligands en compétition. Les ligands peuvent être agonistes : ces molécules ont deux propriétés, celle de se fixer au récepteur et celle de déclencher l’activité associée au récepteur. Les ligands peuvent être antagonistes : ceux-ci n’ont que la propriété de se lier au récepteur, sans déclencher de réponse. LES PROPRIETES DE LA LIAISON SPECIFIQUE : -rapide (Ka). -réversible (Kd). -saturable (KB). Plus une mole de ligand active des récepteurs, lorsqu’elle est en basse concentration, plus le ligand aura d’affinité avec le récepteur. 15 LES LIMITES : Parfois il est difficile de marquer un ligand, parce que ça risque de lui faire perdre son activité. Il est difficile d’étudier un ligand lorsque son affinité est trop faible. Si on a trop peu de récepteurs par rapport au ligand, les récepteurs vont vite être saturés et les ligands en excès vont se lier de façon non-spécifique, ailleurs que sur les récepteurs (membranes plasmiques…). Si on marque différents ligands et qu’on les étudie, on ne peut pas savoir s’ils sont antagoniste ou agonistes (on étudie la formation de la liaison ligand/récepteur, pas l’activité qui en découle). APPLICATIONS. On peut réaliser une expérience de photo-affinité bi-fonctionnelle on rend covalente la liaison, donc irréversible. On peut ensuite faire un SDS-PAGE pour mesurer les poids moléculaires. On cherche sur notre gel les bandes radioactives. Mais cela ne règle pas le problème des liaisons non-spécifiques. On est donc obligé des réaliser une expérience avec un excès de ligands non-marqués et on fait la différence (comme l’expérience précédente. En ce qui concerne la localisation, on met des ligands radioactifs, on les mets en présence de différentes membranes cellulaires. On fait notre photo-affinité bi-fonctionnelle, on fait migrer et on réalise une autoradiographie. On regarde sur quelle types de membranes cellulaires on a de la radioactivité (par exemple : que sur les neurones…). E XEMPLE DU LYSOZYME . Le lysozyme, l’enzyme force le substrat à adopter une config particulière pour catalyser la lyse. C’est Flemming qui a découvert le lysozyme. 2tant malade, il a fait tomber un peu de mucus nasal dans une boite de culture, ce qui a entrainé la mort de bactérie en culture. Il a découvert que c’était le lysozyme qui tuait les bactéries. En coupant la partie polysaccharidique des parois cellulaires de bactérie, ce qui les fait exploser. Cette partie est un polymère de NAG et de NAM. Le lysozyme coupe après un NAM, jamais après un NAM (jamais après un NAG). Ainsi le lysozyme peut attaquer les chaines NAGNAM et les chaines NAG-NAG. On trouve beaucoup de lysozyme dans les larmes pour protéger les yeux. Mais surtout dans le blanc d’œuf de poule. Seulement 129 acides aminés avec 4 ponts disulfures (ce qui est beaucoup en proportion). C’est une structure rigide. 14,6 kDa. On y trouve des hélices alpha et des feuillets bêta. Structure globulaire. Il a été difficile de localiser le site actif de la molécule. On a donc associé cette enzyme à un inhibiteur compétitif, ou à un très mauvais substrat (ce qui revient au même). On a donc choisit un trimère (NAG)x3, on repère où se fixe ce très très mauvais substrat, et on repère ainsi le site actif. On se rend compte qu’il y a de la place pour 16 trois trimère de NAG. Plus on met de NAG, plus la vitesse de catalyse est rapide. On atteint la vitesse maximale à 6 NAG. On remarque que 5 NAG rentrent parfaitement dans une sorte de crevasse structurelle du lysozyme. Le 6ème ne peut rentrer que sous forme demi-chaise, ce qui est thermodynamiquement très peu stable pour le cycle de NAG. Dans les 6 NAG qui rentrent dans le site, le lysozyme coupe la liaison entre le 4 et le 5 (ce n’est ni les liaisons 1-2 ni 2-3, ça peut pas être la liaison 3-4 ni 5-6 parce qu’on n’arrive pas mettre de NAM en 3 ni en 5 par encombrement stérique). La lyse se fait avant le O du 5ème NAG. De part et d’autre de la liaison, on trouve des chaines latérales à carboxylate : Asp en position 52 et Glu en position 35. Le problème du Glutamate35 est dans un milieu hydrophobe, il sera donc moins enclin à céder sont proton. Ce proton va finalement aller sur le O de la liaison entre de NA, C + est stabilisé par la charge – de l’aspartate déprotoné. Une molécule d’eau donne un proton au Glutamate et un OH - au carbone +. L’ion carbonium formé pendant un instant est l’état de transition : l’état de plus haute énergie de la catalyse. On appelle ce genre de mécanisme les catalyses générales acides (à savoir qu’il existe aussi les catalyses générales basiques). L’enzyme va forcer le cycle D à se mettre sous forme demi-chaise. Ce qui permet de répartir la charge positive de l’ion carbonium sur 4 atomes de carbone lors de la catalyse. Expérimentalement on crée un polymère NAG, ou le 4 ème NAG est directement sous forme demi-chaise, en rajoutant un atome d’oxygène au bon endroit : on obtient un (NAG)3-NAGlactone. On remarque que cette molécule est beaucoup plus facilement catalysée par le lysozyme. L’hydrolyse est catalysée de manière optimale à pH 5. A 5 l’asp 52 est déprotoné et le glu35 est toujours protoné. Le mécanisme de transglycosylation. Si on fait une hydrolyse du (NAG) 4 (hydrolysé très lentement), on obtient du (NAG)2 et étrangement, du (NAG)6. Cela s’explique par le fait qu’en présence d’une haute concentration en (NAG)4, le (NAG)2 s’y lie pour former du (NAG)6. E XEMPLE DE LA CARBOXYPEPTIDASE . Protéine de 307 acides aminés de 34,4 kDa. Contenant un seul pont disulfure elle est beaucoup plus souple que le lysozyme. Protéine globulaire contenant des hélices α et des feuillets β. On a comme caractéristique un atome de zinc facilement repérable. L’atome de zinc se trouve dans le site actif et y est très fortement lié. On connait un très très mauvais substrat : le dipeptide Gly-Tyr. Les 5 interactions du dipeptide : -liaisons covalente entre GY et la carboxypeptidase. -liaison hydrogène entre GY et la carboxypeptidase. -l‘extrémité Y entre dans une poche apolaire de l’enzyme. -le zinc hyperpolarise la liaison covalente dans le dipeptide. -l’extrémité N-term fait une liaison hydrogène avec le Glu 270 de l’enzyme. Ce Glu joue un rôle clé dans le mécanisme catalytique voilà pourquoi GY est un mauvais substrat de la carboxypeptidase. 17 Avec un « bon substrat », on n’a que les 4 premières phases qui ont lieux. Le Glu 270 est libre et peut participer efficacement à la catalyse. La charge négative du Glu 270 vient attaquer le carbone de la liaison peptidique déjà très polarisée, hyperpolarisée par le zinc et par l’Arg 127. Une molécule d’eau s’intercale dans la liaison peptidique très fragilisée : c’est la lyse ! On s’est aperçu que la liaison au substrat modifie la structure de l’enzyme. C’est le substrat qui induit la formation du site catalytique. C’est le modèle d’ajustement induit ou ajustfit. Merci à MoD pour les compléments de cours. Méta-Zoheir. 18