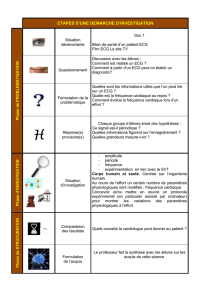
Anomalie de conduction dans une population pédiatrique de une

Anomalie de conduction dans Anomalie de conduction dans
une population pédiatrique deune population pédiatrique de
une
population
pédiatrique
de
une
population
pédiatrique
de
dystrophie myotonique de dystrophie myotonique de
St i tSt i t
St
e
i
ner
tSt
e
i
ner
t
Anne Fournier, mdAnne Fournier, md
Cardiolo
g
ue
p
édiatreCardiolo
g
ue
p
édiatre
gpgp
Lucie Caron, infLucie Caron, inf
Clinique des maladies neuroClinique des maladies neuro--musculairesmusculaires
Centre de réadpatation MarieCentre de réadpatation Marie
--
EnfantEnfant
Centre
de
réadpatation
MarieCentre
de
réadpatation
Marie
EnfantEnfant

Anomalie de conduction dans Anomalie de conduction dans
lti éditi dlti éditi d
une popu
l
a
ti
on p
édi
a
t
r
i
que
d
e une popu
l
a
ti
on p
édi
a
t
r
i
que
d
e
dystrophie myotonique de dystrophie myotonique de
SteinertSteinert
Mise en contexteMise en contexte
Atteinte cardia
q
ueAtteinte cardia
q
ue
qq
–– Anomalies de conductionAnomalies de conduction
––
A
utres anomalies
A
utres anomalies
Population pédiatriquePopulation pédiatrique

D
y
stro
p
hie m
y
otoni
q
ue D
y
stro
p
hie m
y
otoni
q
ue
yp y qyp y q
de Steinertde Steinert
Maladie neuromusculaire héréditaire la plus Maladie neuromusculaire héréditaire la plus
fréquentefréquente
1/8000 naissances1/8000 naissances
1/8000
naissances1/8000
naissances
2.1 2.1 –– 14.3/100 000, 14.3/100 000, > canadiens français> canadiens français
2formes:2formes:
2
formes:2
formes:
–– DM1: maladie de SteinertDM1: maladie de Steinert
DM2 f f t d DM1DM2 f f t d DM1
––
DM2
:
f
orme
f
rus
t
re
d
e
DM1DM2
:
f
orme
f
rus
t
re
d
e
DM1

D
y
stro
p
hie m
y
otoni
q
ue D
y
stro
p
hie m
y
otoni
q
ue
yp y qyp y q
de Steinertde Steinert
A
utosomal dominant
A
utosomal dominant
Anticipation génique (maternelle)Anticipation génique (maternelle)
DM1 Ré étiti CTG h 19DM1 Ré étiti CTG h 19
DM1
:
Ré
p
étiti
on
CTG
c
h
romosome
19
DM1
:
Ré
p
étiti
on
CTG
c
h
romosome
19
(légere: 50(légere: 50--99 répét., classique:10099 répét., classique:100--750 750
répét., congénitale: répét., congénitale: > 750 répét.> 750 répét.))
DM2: DM2: répétition CCTG chromosome 3répétition CCTG chromosome 3
Nombre de répétitions corrèle avec atteinte Nombre de répétitions corrèle avec atteinte
neuroneuro
musculaire et cardiaque chez DM1musculaire et cardiaque chez DM1
neuroneuro
--
musculaire
et
cardiaque
chez
DM1musculaire
et
cardiaque
chez
DM1

D
y
stro
p
hie m
y
otoni
q
ue D
y
stro
p
hie m
y
otoni
q
ue
yp y qyp y q
de Steinertde Steinert
Atteinte multisystémiqueAtteinte multisystémique
–– Faiblesse musculaire, douleurs musculaires, myotonieFaiblesse musculaire, douleurs musculaires, myotonie
Atteinte gastroAtteinte gastro
intestinale: lithiase biliaire dysphagieintestinale: lithiase biliaire dysphagie
––
Atteinte
gastroAtteinte
gastro
--
intestinale:
lithiase
biliaire
,
dysphagieintestinale:
lithiase
biliaire
,
dysphagie
–– CataractesCataractes
–– Apnées du sommeilApnées du sommeil
–– Infertilité Infertilité -- Résistance à lRésistance à l’’insuline insuline -- Calvitie frontaleCalvitie frontale
–– Somnolence diurneSomnolence diurne
––
Retard mentalRetard mental
Retard
mentalRetard
mental
–– Atteinte cardiaqueAtteinte cardiaque
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
1
/
48
100%