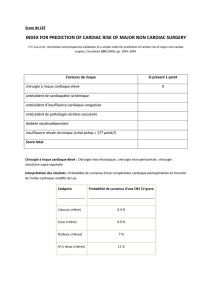
Hélène Puccio - MeetOchondrie

Mieux comprendre l’ataxie de Friedreich par le
développement de modèles
cellulaires et murins
Hélène PUCCIO
Institut de Génétique et de Biologie
Moléculaire et Cellulaire
(IGBMC)

Transmission Autosomique Recessive
- prévalence 1:50000 : la plus fréquente des ataxies héréditaires
- apparition des symptômes < 25 years
Friedreich Ataxia: une maladie mitochondriale
Signes extra-neurologiques
- Cardiomyopathie hypertrophique
- Diabète ou intolerance au glucose
Membres
inférieurs
Membres
supérieurs
Extrémité
céphalique
Instabilité
marche
Dysarthrie
Troubles déglutition
Maladie neurodégénérative progressive
- Atteinte combinée SNC et SNP
- Troubles de l’équilibre
- Ataxie spinocérébelleuse: perte de la coordination motrice
- Abolition des reflexes ostéo-tendineux
- Syndrome pyramidal
perte des capacités ambulatoires

Cortex
Cerebellum
Medulla
Spinal
cord White matter
Grey matter
Clark
Column
Pyramidal tracts
Posterior
column
Sensory
neurons
Spinocerebellar
tracts
Thalamus
Motor neuron
Dorsal root ganglia
Pyramidal
signs
Contrôle
N
N
N
N
Atteinte du SNC dans FRDA

Physiopathologie de FRDA
Accumulation de fer: dans le cœur et le noyau dentelé
Photo: Dr. A Koeppen
Déficit des enzymes Fe-S: CI-CIII de la chaîne respiratoire + aconitases
Stress oxydant: augmentation des marqueurs de stress oxydant
dans les urines et le sang
Physiopathologie: hypothèses jusqu’en 2001
Frataxine Fer Stress oxydant Enzymes mitochondriales (Fe-S)

FRDA: le gène de la frataxine
Gène FXN (ou FRDA) en 9q21.11 (1996)
ARNm
Protéine FRATAXINE (FXN)
- cœur, moelle épinière +++
- pancréas, foie, muscles squelettiques, cervelet ++
- signal d’adressage mitochondrial
- protéine très conservée
- sans similitude avec une protéine de fonction connue
ADN
Protéine mature
Domaine hautement conserv é
1
ATG
210 aa
1 2 3 5a4
1,3 kb
1,3 kb
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
1
/
38
100%