Les aminoacidopathies
Année 2018/2019 Dr ARAB
Biochimie clinique

Biochimie clinique
Les aminoacidopathies
2
Objectifs :
A l’issu de ce cours, le résident doit être capable de :
- Lister les voies de dégradation de la chaine carbonée et du groupement aminé des
acides aminés
- citer les enzymes régulant le cycle de l’urée
- Décrire les enzymes déficitaires ou mutées dans les aminoacidopathies et voies
d’activation secondaires
- Différencier et interpréter le diagnostic biologique des aminoacidopathies
- Donner les complications des aminoacidopathies en l’absence de traitement
- Définir le traitement nutritionnel et thérapeutique
Plan du cours :
1-Introduction
2- Rappel structural des acides aminés
3- voies de dégradation des acides aminés
3-1 Voie de dégradation du groupement aminé des acides aminés
3-2 Voie de dégradation de la chaine carbonée des acides aminés
3-3 métabolismes de certains acides aminés :
3 3-1 méthionine
3-3-2 phénylalanine et tyrosine
4- Les aminoacidopathies :
4-1 méthodes d’exploration des aminoacidopathies
4-2 La phenycetonurie
4-3 désordres du métabolisme de la tyrosine
4-3-1 tyrosinemie type I
4-3-2 tyrosinémine type II
4-3-3 tyrosinémie type III
4-3-4 Alcaptonurie
4-4 La leucinose
4-5 Acidurie organique
4-6 Homocystinurie
4-7 cystinose
4-8 cystinurie
4-9 hyperamminièmie

Biochimie clinique
Les aminoacidopathies
3
1- Introduction :
Les maladies héréditaires du métabolisme (MHM) résultent le plus souvent d’un déficit
enzymatique sur l’une des nombreuses voies métaboliques, dérivées des glucides, protides ou
acides gras ou du trafic intracellulaire.
Un déficit enzymatique entraine l’absence d’un compose situe en aval du déficit et/ou
l’accumulation d’un composé potentiellement toxique situé en amont du déficit. Il se
manifeste à tout âge de la vie, le plus souvent en période néonatale mais aussi pendant
l’enfance ou à l’âge adulte lorsqu’il s’agit de déficits enzymatiques partiels. Ils sont encore
insuffisamment diagnostiqués, en particulier en pathologie adulte où les déficits enzymatiques
partiels peuvent se manifester par des tableaux cliniques très varies, notamment des
manifestations digestives, des épisodes psychiatriques ou un retard mental.
Une fois ingérées, les protéines sont absorbées au niveau du tube digestif sous forme d’acides
amines (AA) et peuvent ainsi circuler dans le sang. Les AA sont dégradés en acides
organiques (AO), indétectables chez le sujet sain dans le plasma ou faiblement excrétés dans
les urines. Ces AO sont essentiels au cycle de Krebs. Cette dégradation peut se faire grâce à
des outils : les enzymes. Un déficit enzymatique entraine l’accumulation des AA ou des AO
concernés par l’enzyme déficitaire, ou l’accumulation de substances toxiques comme
l’ammoniac (NH3).
Les AA proviennent des protéines alimentaires (intoxication exogène). Les maladies
d’intoxication les plus fréquentes sont les aminoacidopathies, principalement la leucinose, la
phenylcetonurie et la tyrosinemie de type I, les aciduries organiques dont l’acidémie
methylmalonique, l’acidémie propionique et l’acidémie isovalerique, et enfin les déficits du
cycle de l’urée.
les anomalies du métabolisme des folates et des cobalamines,l’homocystinurie, l’intolerance
aux protéines dibasiques et le syndrome « HHH»
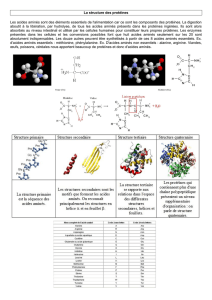
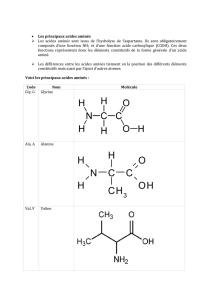
2-Rappel structural des acides aminés :
La plupart des protéines contiennent dans des proportions variables les 20 acides
aminés α L .
Il existe 10 acides aminés dits indispensables ou essentiels apportés par l’alimentation où
l’organisme est incapable de les synthétiser et 10 acides aminés dits non essentiels ou non
indispensables. Les AA ont un motif structural commun

Biochimie clinique
Les aminoacidopathies
4
Acide aminé essentiels: la valine, la leucine, l’isoleucine, la phénylalanine, le tryptophane, la
lysine, la méthionine et la thréonine : Lysine et Thréonine: Absolument essentiels.
Acides aminés parfois essentiels, dans certaines conditions (grossesse, croissance) l’histidine
et l’arginine deviennent essentielles ; (Arg +++ = chez le Nourrisson)
Les acides aminés qu’on peut appeler semi essentiels : tyrosine et la cystéine (phénylalanines
et la méthionine)
3-voies de dégradation des acides aminés :
Le catabolisme ou dégradation des acides aminés s’accompagne toujours de l’enlèvement de
l’azote aminé (c’est la 1ère réaction catabolique).
Le squelette carboné restant, appelé acide α-cétonique (ce n’est pas un AA), est à son tour
dégradé en intermédiaires.
3-1 élimination du groupement aminé :
Différentes voies d’élimination :
- Transamination: commune à tous les AA sauf la lysine.
- Désamination oxydative: le glutamate.
- Désamination non oxydative: la sérine, cystéine et la thréonine.
Cela conduit à la production d’un composé toxique pour le système nerveux central:
l’ammoniac (NH3). Celui-ci est éliminé (systèmes de détoxication) de l’organisme
Sous forme d’urée (uréogénèse= voie majeure hépatique qui représente 4/5 de l’azote éliminé)
ou sous forme de NH4+ (l’ammoniogénèse rénale= forme mineure
L'ammoniac NH3 est un composé toxique. Il est formé dans les tissus périphériques (et le
foie) à partir des AA par une série de réactions de transamination et de désamination. Il est
également produit par les bactéries dans l'intestin.
Il se déplace vers le foie et vers le rein, principalement sous la forme de glutamine (et
d’alanine) pour être éliminé.
Le transporteur d’azote entre les différents organes = glutamine.
Synthèse de la glutamine (Glutaminogénèse)
Se déroule au niveau des tissus périphérique
C’est la synthèse de la glutamine à partie du glutamate via la glutamine synthétase
cytosolique
Hydrolyse de la glutamine :

Biochimie clinique
Les aminoacidopathies
5
Figure1 : glutamine transporteur de l’ammoniac
L’ammoniogénèse :
Dans le rein, le NH3 libéré à partir de la glutamine va s’associé avec des H+ pour former l’ion
ammonium (NH4+) qui sera éliminé dans les urines.
Figure 2 : schéma de l’ammoniogénèse
Le cycle de l’urée ou uréogenèse :
Au niveau du foie, le NH3 libéré à partir de la glutamine est pris en charge par le cycle de
l’urée = uréogenèse.
Le cycle de l’urée est la voie préférentielle de l’élimination de l’azote en excès.
L’urée n’a aucune fonction physiologique.
C’est un cycle qui fait intervenir en particulier les AA suivants: Arginine, Ornithine,
Citrulline et l’Argininosuccinate.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
1
/
21
100%