Transmission des maladies génétiques : Cours de génétique
Telechargé par
angele.tengo

Transmission des maladies génétiques
Maladie congénitale : arrive à la naissance. Maladie acquise : cause extérieur (infection,
trauma, accident). Maladie génétique. Une maladie génétique peut ne pas être héréditaire
quand les mutations apparaissent dans les cellules somatiques (non transmises à la
descendance). La maladie génétique peut être monogénique/monofactorielle (causée par un
seul gène).
Mode de transmission maladie génétique monofactorielle suit lois de Mendel. D’où maladie
mendélienne. Si même gène impliqué chez les patients, maladie mendélienne non
hétérogène.
NB : Lois de MENDEL. 1ère : Uniformité des hybrides de première génération. 2ème : Loi de
pureté des gamètes. 3ème : Ségrégation indépendante des caractères.
Les lois de Mendel sont à la base de l'explication des modèles d'héritage des traits
génétiques des êtres vivants.
Quatre modes de transmission : autosomique ou lié à l’X, dominant ou récessif.
Pénétrance : probabilité d’être atteint sachant le génotype. Quand il y a pénétrance
incomplète, on peut avoir le génotype à risque sans être atteint de la maladie.
Expressivité variable : pour un même génotype à risque, la maladie peut prendre plusieurs
formes.
Empreinte parentale : expression monoallélique d’un gène selon origine parentale.
Hérédité mitochondriale : maladies à transmission maternelle exclusive (mode de transmission
non mendélien).
Hérédité multifactorielle : dépend de l’environnement et de la génétique. Combinaison
particulière d’allèles normaux de certains gènes qui est pathologique.
Mode autosomique dominant : gène morbide porté par un autosome, suffit d’une seule copie
pour être malade. Les individus homozygotes pour l’allèle morbide sont rares. A chaque
grossesse, le risque que l’enfant soit malade est de 50%. 1 risque sur deux de transmettre
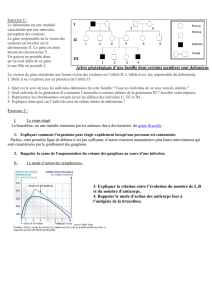
l’allèle morbide. Une personne malade a un de ses deux parents atteints.
Transmission des maladies autosomiques dominantes s’effectue sans saut de génération si
pénétrance complète. TRANSMISSION VERTICALE
NB : en toute rigueur, l’individu malade ne transmet pas sa maladie mais l’allèle muté qui ne
peut être contrecarré par l’autre parent.
Quand les deux parents sont atteints (par exemple dans les maladies relativement fréquentes
comme les hypercholestérolémies familiales).
Faire tableau des gamètes de la méiose des parents.
Un sujet homozygote pour le caractère dominant peut avoir une atteinte plus sévère, plus
précoce ou plus rapidement évolutive.
Dans les modes autosomiques dominant, la personne est forcément atteinte. Pas de
porteurs sains. Les individus sains ont 0% de chance de transmettre l’allèle muté.
Particularité : mutation de novo peut se produire dans cellules germinales de l’un des parents.
Le parent ne sera pas atteint mais peut transmettre l’allèle muté à sa descendance. Dans ce
cas, pas d’histoire familiale. Exemple : achondroplasie ou syndrome de Marfan, taux de
néomutations est élevé. Certaines néomutations favorisées par un âge paternel avancé.
Comment détecter les cas de néomutations ? Quand l’individu malade a des parents sains
mais des enfants qui sont malades. Une mutation qui arrive de manière sporadique dans la
population.
Particularité : pénétrance incomplète.
Probabilité d’être atteint par la maladie quand on a le génotype à risque. Pénétrance d’une
maladie complète (1) quand tous les individus porteurs de l’allèle muté (génotype à risque)
sont malades. Pénétrance incomplète : quand les individus porteurs du génotype à risque ne

sont pas forcément malades. Pénétrance peut être incomplète si la maladie dépend de
l’environnement, autre gène, expression inégale des deux copies d’un gène. NB : les
syndromes de prédisposition au cancer fréquemment une pénétrance incomplète.
Caractéristiques : il peut y avoir des sauts de générations, ressemble à un mode de
transmission autosomique récessif. Dans le cas du rétinoblastome, on a un saut de génération
malgré que l’allèle pathologique soit présent à toutes les générations. Observations conformes
au mode autosomique dominant avec pénétrance incomplète.
Dans le rétinoblastome, maladie autosomique dominant avec une pénétrance de 90%.
Chaque individu porteur de l’allèle pathologique a un risque de 50% de transmettre la maladie.
90% des individus portant l’allèle muté seront malades.
Particularité : expressivité variable
Dans une même famille, des personnes ayant hérité de la même mutation, peuvent présenter
des symptômes cliniques différents. Surtout observé dans les maladies dominantes. Certains
porteurs de l’allèle muté peuvent n’avoir que des signes bénins. La transmission semble alors
sauter une génération. Ils ne seront pas considérés comme cliniquement malades.
Maladies autosomiques récessives
Si le gène en cause est porté par un autosome et si la présence de deux allèles mutés du
gène est nécessaire pour que la maladie se manifeste. Les malades sont homozygotes pour
le gène en cause. Autant de filles et de garçons qui sont touchés car le gène impliqué est porté
sur un autosome. Il n’y a pas de personnes malades à toutes les générations, généralement
les sujets naissent de parents hétérozygotes. Aucun des parents des individus malades ne
sont atteints.
La maladie du fait des faibles dimensions des familles humaines peut toucher qu’une seule
personne dans la famille. Cas isolé ne signifie pas cas de novo. Risque que l’enfant soit
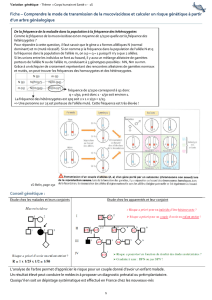
malade est de 25%. Faire tableau des gamètes de la méiose des parents.
Le risque pour un couple d’avoir un enfant atteint d’une maladie récessive dépend du risque
pour chaque conjoint d’être hétérozygote. Lié à la fréquence des hétérozygotes dans la
population.
Si maladie congénitale, fréquence de la maladie peut être donnée par sa prévalence à la
naissance.
Cas de la consanguinité
Enfants qui résultent d’unions entre apparentés (individus qui ont au moins un ancêtre
commun). Conséquence fréquente des mariages consanguins : favorise chez les enfants
consanguins la réunion de deux allèles pathologiques venant d’un ancêtre commun,
responsable d’une maladie récessive. Risque d’autant plus grand que les conjoints sont
proches parents. Plus la maladie est rare, plus l’accroissement du risque dû à la consanguinité
est accru. Si les deux conjoints ont reçu l’allèle pathologique, ils peuvent le transmettre à leur
descendant qui ayant les deux copies de l’allèle pathologique sera malade. Risque de ¼ pour
l’enfant d’être atteint. Dans population où nombreux mariages consanguins, on observe une
augmentation de la fréquence des enfants atteints de maladie récessive. La consanguinité
n’affecte pas la fréquence de l’allèle pathologique mais favorise seulement leur présence en
double exemplaire chez les enfants consanguins.
Risques pour la descendance. La maladie se transmet selon un mode autosomique récessif
selon des règles identiques qui s’apparentent à aux couples non apparentés. La consanguinité
ne modifie pas la fréquence de l’allèle pathologique.
Les augmentations de risque sont d’autant plus fortes que la maladie est rare et que la parenté
est forte. Les ancêtres communs ne peuvent être porteurs de tous les allèles pathologiques
du génome.

Cas particulier pseudodominance. Quand la fréquence des hétérozygotes est importante au
sein de la population, qui fait que la maladie est présente à chaque génération. Exemple :
drépanocytose fréquente en Afrique (sélectionné car résiste au paludisme). La probabilité de
rencontrer un hétérozygote en se mariant au hasard dans la population est élevée.
Cas de l’hétérogénéité génétique
Hétérogénéité intragénique : existence pour un même gène de plusieurs allèles mutés
différemment mais ayant la même conséquence pathologique. Hétérozygote composite.
Hétérogénéité intergénique : des mutations dans des gènes différents peuvent conduire au
même phénotype malade.
Dans ces deux conditions, deux individus atteints de la même maladie peuvent être touchés
dans deux gènes différents mais aussi dans le même gène.
Maladie monofactorielle : un seul gène mis en jeu dans la maladie mais pas le même d’un
patient à l’autre.
Maladies dominantes liées à l’X
Individus de sexe masculin n’ont qu’un seul exemplaire du chromosome X : ils sont
hémizygotes et ne possèdent qu’un seul exemplaire des gènes du chromosome X. Les
femmes possèdent deux chromosomes X. Chez les individus masculins, la question de
dominance ou de récessivité ne se pose pas. Soit le gène est muté, les hommes sont atteints,
soit le gène est normal, les hommes sont sains.
Cette question ne se pose que chez les femmes. Si la maladie survient quand un seul des
gènes est muté, elle est dominante. Sinon (quand les deux exemplaires sont mutés),
récessive.
Les deux sexes peuvent être touchés. Les filles hétérozygotes sont moins sévèrement
touchées que les garçons hémizygotes. Il y a plus de femmes atteintes que d’hommes.
Logique ? Quand un homme est malade, il transmet son X malade à toutes ses filles mais pas
à ses fils. Transmission verticale. Il n’y a jamais de transmission père-fils.
Risques de transmission différents selon que ce soit la mère ou le père atteint. Si le père est
atteint, toutes ses filles seront atteintes et aucun de ses fils. Si la mère est atteinte, 50% de
risque que la fille/fils soit atteint.
Cas d’un père et d’une mère malades : toutes les filles auront l’allèle X malade. Celles qui ont
reçu les deux exemplaires du chromosome muté transmettront la maladie à tous leurs enfants.
50% que le garçon soit malade (reçu l’allèle X muté) de sa mère et les filles sont toutes
malades car elles ont reçu l’allèle muté de leur père et de leur mère.
Dans les cas du X dominant, le père malade aura tous ses filles malades et les mères malades
auront la probabilité de transmettre l’allèle muté à leurs fils de 50%. Rappel : pas de
transmission père-fils.
Inactivation du chromosome X chez les femmes
Dans chaque cellule d’une femme, un des X est inactivé (aléatoirement, très tôt dans le
développement). Le mosaïsme n’affecte que le chromosome X. Les autres chromosomes sont
identiques dans les deux types cellulaires.
Si chez une femme hétérozygote, l’inactivation touche le X porteur du gène normal, la cellule
n’exprimera que le gène muté. Si inverse, seul le gène normal sera exprimé par la cellule. Les
femmes hétérozygotes pour le gène impliqué dans une maladie dominante liée à l’X plus ou
moins atteinte selon le X inactivé.
On peut avoir des maladies liées à l’X dominant léthales pour les fœtus de sexe masculin car
l’absence d’un allèle normal ne peut fournir une protéine fonctionnelle.
Maladies récessives liées à l’X
Ici pareil, chez les individus masculins, la question de dominance-récessivité ne se pose pas.
Si le gène est muté, hommes malades. Si le gène est normal, hommes sains. Dans les cas de
femmes, si la maladie survient quand les deux X sont mutés, la maladie est récessive.
Risques de transmission diffèrent selon que la mère ou le père soit atteint.

Si le père est atteint et que la mère est hétérozygote saine, 50% des fils sont malades et 50%
des filles sont malades.
Si la mère est atteinte et pas le père. Si la mère est malade, tous ses fils sont malades.
Toutes les filles d’un père malade seront conductrices.
L’empreinte parentale est une inactivation sélective de quelques gènes, durant la
spermatogénèse ou l’ovogénèse, de sorte que le génome issu de la fécondation est
fonctionnellement haploïde pour ces gènes (haploïdie fonctionnelle). Pour les gènes soumis à
l’empreinte (inactivation) paternelle, seul l’allèle maternel est exprimé. Pour les gènes soumis
à l’empreinte (inactivation) maternelle, seul l’allèle paternel est exprimé. L’empreinte a lieu au
cours de la formation des cellules sexuelles. Refaite avant chaque fécondation.
Absence d’altération de la séquence des gènes. Réversibilité à chaque génération.
Implications
Toute anomalie génétique qui empêchera la transcription de cet exemplaire unique entrainera
la suppression totale de l’expression du gène concerné.
Mode de transmission des phénotypes dépend des gènes sous empreintes parentales, aussi
du sexe de l’enfant.
Imprinting : methylation to turn off genes. Depends on maternal vs paternal inheritance.
High yield diseases : Prader-Willi Syndrome/Angelman Syndrome.
1
/
4
100%