Pharmacocinétique : Distribution des médicaments - Cours de médecine
Telechargé par
Nabil Belabbes

O
Or
rd
do
on
nn
na
an
nc
ce
e
e
et
t
r
rè
èg
gl
le
es
s
d
de
e
p
pr
re
es
sc
cr
ri
ip
pt
ti
io
on
n
D
Dr
r:
:
D
DJ
JE
ED
DI
ID
D.
.S
S
Pharmacocinétique
2. DISTRIBUTION
Le Plan
I. Introduction
II. Définition
III. Fixation des médicaments sur les constituants sanguins
1. Sur les éléments figurés du sang
2. Sur les protéines plasmatiques
IV. Fixation dans les tissus
1. Mécanismes de passage
2. Facteurs limitant la diffusion tissulaire des médicaments
V. Passage particulier des médicaments
1. Distribution dans le système nerveux central
2. Diffusion fœtoplacentaire
VI. Notion du volume de distribution
VII. Conclusion
I. Introduction
Après l’étape de résorption, cas d’une administration extravasculaire, le
médicament se retrouve dans la circulation générale. Dans l’espace
vasculaire, les médicaments se fixent sur les protéines plasmatiques par des
liaisons réversibles. La fraction libre du médicament, qui est en équilibre
avec la fraction liée, va quitter la circulation sanguine pour diffuser dans les
tissus et les compartiments liquidiens. Certains médicaments se fixent
également sur les éléments figurés du sang.
II.Définition
La diffusion est la répartition du médicament dans l’ensemble de l’organisme.
Elle se fait en 2 temps :
1/9
3éme année médecine

Le transport du médicament dans le sang.
La diffusion tissulaire c.à.d. le passage du médicament du sang vers les tissus
et organes.
III. Fixation des médicaments sur les constituants du sang
1. Fixation sur les éléments figurés du sang
Moins importante que la fixation sur les protéines plasmatiques.
Intervention d’hématie: membrane cellulaire ou les constituants intracellulaires
(hémoglobine et l’anhydrase carbonique).
Pentazocine, Promazine, Salicylate
La valeur des paramètres pharmacocinétiques, des médicaments qui se fixent sur les
éléments figurés du sang, doit être exprimée en données sanguines et non
plasmatiques.
Le rapport érythroplasmique mesure l’importance de la liaison [hématie –
médicament].
La régulation du pH sanguin et le transport du CO2 grâce à l'anhydrase carbonique,
une enzyme présente à la surface des hématies qui transforme les bicarbonates en
CO2ou l'inverse, selon les besoins du corps. Ainsi, les hématies transforment le
CO2 fabriqué par les cellules en bicarbonates, puis elles vont jusqu'aux poumons, où
2/9

elles retransforment le bicarbonate en CO2.
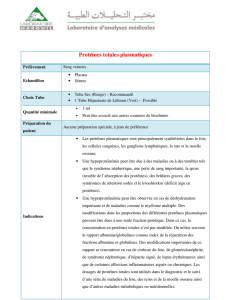
2. Fixation sur les protéines plasmatiques
Les protéines plasmatiques fixant les médicaments:
Nombreuses protéines dans le plasma :
L’albumine est la plus importante sur le plan quantitatif (Représente 50 à 68 % des
protéines du plasma), et fixe un très grand nombre de médicaments, de façon non
spécifique.
L
L’
’a
al
lb
bu
um
mi
in
ne
e:
:
Représente 50 à 68 % des protéines du plasma
Fixe préférentiellement les médicaments à caractère acide faible ( Ex: acide
valproïque, warfarine….)
L’affinité est élevée.
Le nombre de sites de fixation des acides faible sur l’albumine est faible.
Existence de phénomène de saturation; interaction possible.
Les médicaments bases faibles:
Ex : Propranolol , Diltiazem , Rifampicine
se fixe sur:
L’ α1-glycoprotéine acide
Les globulines (α, β, γ)
Les lipoprotéines (HDL ; LDL; VLDL)
L‘albumine: Faible affinité et le nombre de site est élevé,
Pas de phénomènes de saturation, peu d’interaction
Caractéristiques de la fixation protéique des médicaments:
La fixation des médicaments sur les protéines plasmatiques est un phénomène
réversible qui répond à la loi d’action de masse. Cette fixation dépend de la
concentration de la protéine liante ainsi que de l’affinité du médicament pour cette
protéine.
3/9

La forme liée (forme de transport et de réserve) est inactive pharmacologiquement et
ne peut diffuser pour atteindre son lieu d’action. Cette inactivité n’est que
temporaire car les formes liée et libre sont en équilibre réversible. Au fur et à
mesure de la disparition de la forme libre (par diffusion vers les tissus ou
élimination), il y a passage de la forme liée vers la forme libre.
Classification des mdts en fonction du % de fixation protéique:
En pratique, la fixation protéique n’est à considérer que si elle est élevée (> 90
%)
et si le médicament a une marge thérapeutique étroite.
Les facteurs influençant la fixation protéique :
4/9

Acides gras libres : diminution de la fixation protéique
plasmatique des médicaments
Bilirubine : diminution de la fixation protéique plasmatique des
médicaments
Exemple d’interaction médicamenteuse
Quand plusieurs médicaments ont une affinité pour les sites de fixation sur
les albumines plasmatiques, une compétition peut avoir lieu entre ces
médicaments.
Soient deux médicaments A et B (avec affinité de A< B pour les albumines
plasmatiques)
L’administration du médicament B peut réduire la fraction fixée et
augmenter la fraction libre dans le plasma du médicament A
Exemple : Déplacement au niveau des protéines du Diazépam par
l’Aspirine
le medt est transporté dans le compartiment sanguin et peut être sous forme libre ou
lié soit aux éléments figurés du sang (c’est-à-dire les cellules sanguines) ou les
proteines plasmatiques circulantes
IV. Fixation dans les tissus (diffusion tissulaire)
Passage de la forme libre du médicament du sang (compartiment central) vers les
tissus et organes (compartiment périphérique) dans lesquels il se fixe.
Mécanismes de passage:
5/9
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%