Thèse : Propriétés diélectriques des matériaux poreux et teneur en eau
Telechargé par
villa rabeh

THESE
présentée par
Tarik ZAKRI
Ingénieur de l’Ecole Nationale Supérieure de Physique de Grenoble - INPG
pour obtenir le titre de Docteur de
l’Institut National Polytechnique de Grenoble
(Arrêté ministeriel du 23 novembre 1988)
Spécialité : Géophysique, Géochimie et Géomécanique
Contribution à l’étude des propriétés
diélectriques de matériaux poreux en vue de
l’estimation de leur teneur en eau : modèles de
mélange et résultats expérimentaux
Soutenue le 10 octobre 1997
Composition du jury :
M. G. Angenieux (Rapporteur)
M. B. E. Clothier (Rapporteur)
M. M. Labeau (Président)
M. J.-P. Laurent
M. M. Vauclin
Thèse préparée au sein du Laboratoire d’étude de Transferts en Hydrologie
et Environnement - UMR 5564 CNRS-INPG-ORSTOM-UJF

Remerciements
Ce travail de thèse a été effectué au sein du Laboratoire d’étude des
Transferts en Hydrologie et Environnement de Grenoble. Il a bénéficié du
soutien financier du “Programme de Recherches en Hydrologie” de
l’INSU, dans le cadre du Projet “Apport de la Géophysique pour l’étude des
circulations de fluides en subsurface” et du Ministère de l’Education
Nationale, de la recherche et de la technologie au travers de ma bourse
MESR.
En premier lieu, je remercie J.-P. Laurent, qui m’a offert
l’opportunité d’être son premier thésard et qui a dirigé et accompagné ce
travail tout au long de ces trois ans.
Mrs G. Angénieux et B. Clothier ont accepté d’être rapporteurs de
cette thèse. Je les remercie sincèrement pour leur lecture critique et
éclairante sur le sujet.
Mr M. Labeau, Professeur à l’INPG, a généreusement accepté de
présider le jury. Qu’il en soit remercié ici, par ces quelques lignes.
Mr M. Vauclin, directeur du LTHE, a été plus qu’un examinateur.
Il a été un soutien et un correcteur indispensable lors de la rédaction du
manuscrit. Qu’il trouve ici toute ma reconnaissance.
Que tous ceux qui ont, de près ou de loin, participé à la réussite de
cette thèse soient aussi remerciés ; les permanents du LTHE, en
particulier ceux de l’équipe TMP, qui ont toujours été à l’écoute des
thésards du labo, les techniciens, Hervé, Jean-Michel, Robert et Stéphane,

pour leur disponibilité. Frédéric, pour la résolution des casses têtes dans
La Recherche (la revue). Les secrétaires, Hélène, Odette et Sylviane, pour
leur gentillesse et leur bonne humeur. Je remercie également Mrs
Apprahamian (Institut Dolomieu), Bochu et Maniguet (CMTC) pour
l’analyse minéralogique des échantillons par diffraction des rayons X.
Enfin, bonne chance à tous les thésards et aux jeunes du labo pour
la suite de leurs travaux. Ceux, d’abord, qui vont bientôt soutenir : Sophie,
Florentina, Luiz, Mariana, Uta et Céline. Allez, vous tenez le bon
bout...Stéphanie, qui peut compter sur moi pour tester ses cornes de
gazelles (même si je prends des risques...). Christian, qui a su me fournir
en aspirine quand j’en avais besoin. La “brasilian team” (l’un, André,
joue au foot, l’autre, Romeu, en parle). Notre soutien à tous, éternellement
jeune, alias “GM”. Soumia (toujours aller la voir avant de partir en week-
end), Anne, Rachel, Alix, Céline, Nawal, Emmanuel, Abou,Jean, Tao,
Torbjörn...et tous les autres, Sophie et Bertrand, Robert et Marie...
A mes amis de la spectro-team, en particulier Cendrine, Magali et
Fred, Boite and Box, Pif et Haud, Dédé,...Pour tous les éclats de rire.
Enfin, merci à Mr Sauvadet pour m’avoir, il y a longtemps, donné le
goût de la Physique.
Et que ceux que j’ai oublié me pardonnent.

Principales notations
Constantes
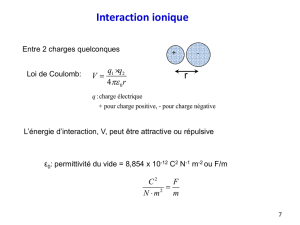
ε0 permittivité diélectrique du vide ou de l’air ( = 8.854 10-12 Farad/m)
µ0 perméabilité magnétique du vide ou de l’air ( = 4π 10-7 Henry/m)
c vitesse de la lumière dans le vide ou dans l’air = 3 108 m/s
i2 = -1
Z0 impédance du câble coaxial = 50 Ω
Grandeurs vectorielles
B
induction magnétique (Vs/m2)
D
Induction électrique (As/m2)
E
champ électrique (V/m)
H
champ magnétique (A/m)
jdensité de courant (A/m2)
r
vecteur position
Grandeurs scalaires
αcoefficient d’atténuation ou affaiblissement linéique
(Np/m)également paramètre du modèle alpha (mais toujours
spécifié dans le texte)
βcoefficient de propagation ou déphasage linéique (rad/m)
χsusceptibilité magnétique ( = 1-µr)
εpermittivité électrique absolue
γcoefficient de propagation : γ = α + iβ
λlongueur d’onde = 2π/β (m)
µperméabilité magnétique absolue (H/m)
µrperméabilité magnétique relative ( = µ /µ0)
θteneur volumique en eau (m3/m3)
θiteneur volumique de la phase i dans un mélange (m3/m3)
ρcoefficient de réflexion (chap. III)
ρamasse volumique du matériau sec dite aussi masse
volumique

apparente (kg/m3)
ρcdensité de charge volumique (As/m3) (chap. I)
ρeau masse volumique de l’eau ( = 1 kg / m3)
ρsmasse volumique de la phase solide (kg / m3)
σconductivité électrique
ωpulsation ω = 2πf
CCapacité (F)
ffacteur de dépolarisation (dans le chap. II)
ffrequence (Hz)
FFacteur de formation
GConductance (Siemens = Ω-1)
Icourant électrique (A)
Kpermittivité électrique relative (=ε/ε0)
K’ partie réelle de la permittivité relative
K0 permittivité du matériau poreux sec ou phase homogénéisée
(solide + air)
-K” partie imaginaire de la permittivité relative
Kb permittivité basse fréquence dans la formule de Debye
K∞permittivité haute fréquence dans la formule de Debye
llongueur (m)
LInductance (H)
ncoefficient de dépolarisation (dans le chap. II)
pporosité
RRésistance (Ω)
Staux de saturation ( = θ/p)
ttemps (s)
vvolume (m3)
Vtension électrique (V)
vvitesse (m/s)
wteneur massique en eau
Yadmittance (Siemens)
Zimpédance caractéristique (Ω)
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
1
/
252
100%