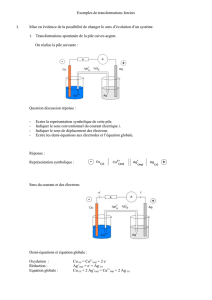
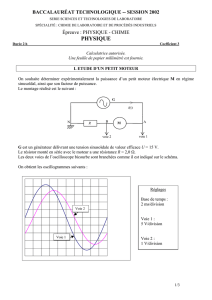
Méthodes électrochimiques de mesure - Cours Master I
Telechargé par
Niamien Paulin Marius

METHODES
ELECTROCHIMIQUES DE
MESURE
Prof. NIAMIEN Paulin Marius, Université FHB (Cocody)
MASTER I Génie des procédés chimiques

Plan du cours
I. Notions d’oxydo-réduction
II. Cinétique des réactions électrochimiques
III. Courbes i=f(E)
IV. Méthodes électrochimiques d’analyse
IV.1 Voltammétrie cyclique
IV.2 Polarographie
IV.3 Ampèrométrie
IV.4 Potentiométrie
IV.5 Coulométrie
2

Définition et Historique
L’électrochimie est la discipline scientifique qui s’intéresse aux relations
entre la chimie et l’électricité. Elle décrit les phénomènes chimiques couplés
àdes échanges réciproques d’énergie électrique.L'électrochimie comprend
toutes technologies et techniques issues de travaux scientifiques liés à
l’électrolyse,la corrosion,les piles,les batteries,l’ électrodéposition, etc.
De plus, l’électrochimie s’intéresse à des systèmes hétérogènes comportant
aux deux extrémités des matériaux conducteurs électroniques (métal,
carbone…) et, entre ces deux conducteurs, au moins un matériau
conducteur ionique (électrolyte liquide ou gélifié, sel fondu…).
Historique
Luigi Galvani (1737-1798): mise en évidence de l’électricité animale
(vidéo).
Alessandro Volta (1745-1827): première pile électrique (empilement
3

de disques métalliques séparés par un tissu ou un carton imbibé d’acide
ou d’eau salée).
Raymond Gaston Planté (1834-1889): invention de la batterie au
plomb(1859)qui sera améliorée plus tard (1881) parAlphonse Faure.
Michael Faraday (1831): principe de l’induction magnétique (création
d’un courant électrique dans un conducteur placé dans un champ
magnétique).
Théophile Gramme (1828-1901): construction de la première dynamo
industrielle en (1871).
On classe les applications industrielles de l’électrochimie en cinq grandes
catégories:
oL’électrosynthèse (aluminium, chlore, soude, lithium, sodium,
magnésium, hydrogène…).
4

oLes traitements de surface et la corrosion : électrodéposition de
métaux(nickel, zinc), anodisation de l’aluminium, l’électrochimie permet
d’expliquer et d’étudier les phénomènes de corrosion;
oLe stockage et la conversion de l’énergie: piles et accumulateurs;
oMéthodes d’analyse et de mesure: conductimétrie, spectroscopie
d’impédance électrochimique, polarographie, méthodes
potentiométriques;
oL’environnement et la biologie: Traitement des eaux, électrodialyse…
5
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
1
/
54
100%