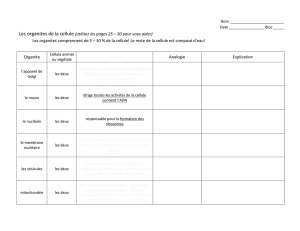
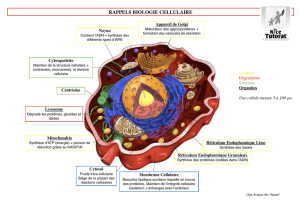
Biologie Cellulaire en 30 Fiches: Manuel de Biologie Cellulaire
Telechargé par
K. Daniel SORO

Comprendre
et s’entraîner
facilement
Jean-Claude CALLEN
Biologie
cellulaire
en 30 fiches


Biologie
cellulaire
en 30 fiches
Jean-Claude CALLEN

© Dunod, Paris, 2009
ISBN 978-2-10-054193-5

© Dunod – La photocopie non autorisée est un délit.
FICHE – 1
Avant-propos
Cet ouvrage est destiné aux étudiants en Biologie (L1 et L2 SV). Comme tous les volu-
mes de cette collection, il vise à leur faire acquérir les bases d’une véritable démarche
expérimentale en Biologie Cellulaire. Longtemps réduite à la seule description des
structures cellulaires : la Cytologie, cette discipline s’est enrichie, au cours du temps,
des méthodes de la Biochimie, de la Biophysique, de la Physiologie Cellulaire et plus
récemment de la Biologie Moléculaire. Il suffit simplement de feuilleter un journal
scientifique dédié à la Biologie Cellulaire moderne pour se convaincre de la multiplicité
des approches qui caractérisent cette discipline.
Afin de coller au plus près à l’actualité de la Recherche, l’enseignement universitaire se
doit donc de former les étudiants aux démarches et aux outils les plus récents ; il est bien
loin le temps où les séances de Travaux Dirigés consistaient à reproduire le plus fidèle-
ment possible des clichés de Microscopie Électronique ! Actuellement, il est indispen-
sable qu’un étudiant en Biologie Cellulaire connaisse un grand nombre de techniques,
de plus en plus sophistiquées. Et si le fractionnement cellulaire et l’électrophorèse ne
doivent plus avoir de secrets pour lui, il lui faut aussi maîtriser les bases de l’immuno-
cytochimie et de l’hybridation in situ, connaître le principe d’un microscope confocal
et d’un trieur de cellules, mais aussi savoir construire un protocole utilisant un système
de traduction in vitro ou la GFP.
C’est dans cet esprit et avec cet objectif que cet ouvrage est construit. Il est divisé en 30
fiches de 5 pages, couvrant 10 grands thèmes classiques de la Biologie Cellulaire. Cha-
cune d’elles inclut une page de rappels de notions indispensables et une page décrivant
une technique précise, en rapport (dans la mesure du possible) avec le sujet de la fiche ;
trois pages sont donc consacrées à des exercices semblables à ceux effectués dans des
Travaux Dirigés. Sur les 62 exercices proposés, près de la moitié consistent en des ana-
lyses d’authentiques expériences de laboratoire (même si certaines sont nécessairement
simplifiées !), avec leurs techniques et leur démarche hypothético-déductive caractéris-
tique. D’autres exercices, reflétant la diversité des outils pédagogiques à notre disposi-
tion, sont proposés à part sensiblement égale : calculs à effectuer, figures à légender,
séries de propositions vraies ou fausses à identifier. Tant il est vrai que, s’il faut savoir
raisonner (ce qui est donné à une majorité d’étudiants !) et exercer son esprit critique
pour réussir en Biologie, il faut également disposer d’une somme toujours plus impor-
tante de connaissances sur lesquelles s’appuyer, et sans lesquelles il n’est hélas pas pos-
sible de progresser.
J.-C. Callen
C’est la profonde ignorance qui inspire le ton dogmatique Jean de La Bruyère
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
1
/
163
100%