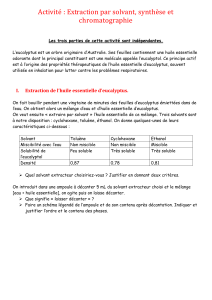
Extraction d'huile essentielle d'Eucalyptus : Méthodes & Analyse
Telechargé par
amy-feghoul

1
INTRODUCTION
De nombreux végétaux contiennent des substances odorante, volatils est peut soluble dans
l’eau appelées « huiles essentielles », celles-ci sont à l’origine de l’odeur parfumé des
plantes. Très vite ces composés ont intéressé les diverses industries, tel que les cosmétiques,
la parfumerie, la pharmacie, l’agroalimentaires etc.… . Ainsi plusieurs techniques ont été
mises au point pour l’isolement de ces constituants selon les caractéristiques de la plante ou
la partie de la plante (feuilles, bois, fruits …).
Quelque soit le secteur d’activité, l’étude des huiles essentielles reste une étape importante
qui, malgré les progrès constants des différentes techniques de séparation et
d’identification, demeure toujours une opération délicate nécessitant la mise en œuvre
simultanée ou successive de diverses techniques
Dans ce travail nous nous somme intéresser à l’extraction de l’huile essentielle des feuilles
d’Eucalyptus globulus par 2 méthode différentes entrainement a la vapeur et
macération(extraction a froid). La caractérisation de ces essences a été faite par la
détermination du rendement et de leurs compositions chimique, donc qualitativement en
utilisant la technique de chromatographie en phase gazeuse en programmation de
température sur une colonne de polarité moyenne, en utilisant le FTIR, la séparation sur
colonne et la CCM.
I.GENERALITEES :
1. Définition :
Telles que définies, les huiles essentielles seraient des produits généralement odorants,
obtenus soit par entraînement à la vapeur d’eau de végétaux ou de parties de végétaux, soit
par expression à froid du. Mais cette définition est très restrictive, car elle exclut d’une part
les produits odorants d’origine animale et d’autre part les essences obtenues par d’autres
procédés d’extraction.
De ce fait quand on examine une huile essentielle il faut avoir à l’esprit deux préoccupations
principales : le matériel botanique d’où elle est issue ainsi que le mode d’extraction.
2. Localisation dans la plante :
Les huiles essentielles sont largement répandues dans le règne végétal, elles peuvent se
rencontrer dans tous les organes végétaux.
Dans une même plante les essences peuvent être présentes à la fois dans différents organes,
leur composition chimique pouvant varier d’un organe à l’autre.

2
3. Modes d’obtention des huiles essentielles :
Au vu de la richesse et de la complexité de leur composition chimique plusieurs techniques
d’extraction de ces essences et des principes aromatiques végétaux ont été mises au point
pour l’exploitation de ces richesses naturelles. Toutefois les normes liées à l’utilisation de ces
essences limitent en général le choix de la méthode d’extraction.
En effet la localisation histologique des composés aromatiques dans le végétal ainsi que la
destination finale du produit extrait peuvent orienter le choix technologique.
Les méthodes d’extraction sont adaptées aux propriétés les plus importantes des huiles
essentielles : leur volatilité dans l’air et dans la vapeur d’eau et leur solubilité dans les
solvants organiques.
Entraînement à la vapeur d’eau :
Ce procédé est basé sur le fait que la plupart des composés odorants volatils (en particulier
les huiles essentielles) contenus dans le végétal sont susceptibles d’être entraînés par la
vapeur d’eau du fait de leur point d’ébullition relativement bas et de leur caractère
hydrophobe. Ils ne sont donc ni retenus (par et dans les biopolymères de la plante), ni
solubilisés dans l’eau. Il en existe deux types :
L’hydrodistillation :
Elle consiste à immerger directement le matériel végétal à traiter (intact ou broyé)
dans un alambic rempli d’eau qui est ensuite porté à ébullition. Les vapeurs
hétérogènes formées sont condensées et l’essence se sépare par différence de
densité.
Distillation à la vapeur d’eau :
Pour ce type de distillation le végétal n’est pas en contact avec l’eau. La vapeur
nécessaire à l’extraction arrive à l’intérieur de l’alambic par le fond et repart de
manière homogène au moyen d’un tuyau circulaire muni de nombreuses ouvertures.
Extraction par solvant organique :
Le matériel végétal est mis en contact avec un solvant à chaud ou à froid (on opère le
plus souvent à température ambiante). Le produit obtenu après évaporation du
solvant est appelé « concrète ». Ce terme résulte de la tendance du produit à se
solidifier en raison de la présence de matière grasse entraînée par le solvant. Le
traitement à froid de la concrète par l’alcool absolu permet ensuite de séparer les
matières grasses et obtenir après évaporation de l’alcool, la phase dite « absolue »
des huiles essentielles.

3
On peut citer également d’autres procédés d’extraction tels :
L’enfleurage ou macération
Extraction au CO2 supercritique
L’extraction par micro-onde.
4. Composition chimique :
La composition chimique d'une Huile Essentielle est complexe et chaque classe chimique est
étroitement liée à une réponse thérapeutique précise.
Ces molécules sont généralement des hydrocarbures terpèniques :
monoterpènes (C10), sesquiterpènes (C15), diterpènes (C20), triterpènes (C30), et
tétraterpènes (C40), tous formés à partir d’un multiple pair ou impair d’unités isopréniques .
Ils ont tous la même origine biogénique : l’isopentényl pyrophosphate, de structure
hémiterpènique qui est le précurseur commun de ces molécules, bien que n’existant pas à
l’état libre dans le végétal.
Les huiles essentielles contiennent surtout des monoterpènes, quelques sesquiterpènes et
plus rarement des diterpènes. Ces terpènes sont prépondérants. Ils peuvent être acycliques,
monocycliques ou bicycliques. Ils sont formés dans cet ordre chronologique dans le végétal
et sont, selon leur degré d’oxydation, fonctionnalisés par des groupes hydroxyles, époxydes,
aldéhydes ou carbonyles.
5. Utilisation des huiles essentielles :
Actuellement les huiles essentielles trouvent leur utilisation dans différents secteurs. Si celui
de la parfumerie est leur débouché principal, ceux des produits d’hygiène et de la
cosmétologie sont également de grands consommateurs, même si le coût souvent élevé des
produits naturels conduit à privilégier les produits synthétiques.
Certaines huiles essentielles sont aussi utilisées en nature dans l’industrie Pharmaceutique
en particulier pour la préparation d’infusion et sous forme de préparation galénique simples.
De plus elles servent à l’aromatisation de certaines formes médicamenteuses destinées à la
voie orale. Les essences naturelles constituent le support d’une thérapeutique particulière :
l’aromathérapie.
6. Les huiles essentielles d’eucalyptus :
Présentation botanique :
Originaire de Tasmanie en Australie, il existe environ 800 espèces dont 50 se sont
acclimatées dans le bassin méditerranéen depuis leur introduction au milieu du 19ème
siècle, ils poussent dans toutes les régions chaudes et tempérées du monde jusqu’à
1000 m d’altitude.

4
Le genre Eucalyptus a été baptisé ainsi en 1788, c’est le terme, dérivé du grec « eu=
bien et kalyptos= couvert » que le botaniste Français L’HERITIER a choisi pour décrire
l’opercule qui recouvre les étamines dans le bouton floral.
L’étude taxonomique décrite par VRODOIJAK (1965) et BECHKOK (4) a permis de
déterminer la systématique suivante :
- Embranchement : Spermatophytes
- Sous-embranchement : Angiospermes
- Classe : Dicotylédones
- Sous-classe : Diatyptales
- Famille : Myrtacées
L’espèce la plus connue de tous est l’Ecalyptus globulus surnommée également «
gommier bleu de Tasmanie » ou encore « arbre de la fièvre » et décrite pour la
première fois par LABILLARDIERE vers 1799-1800 d’où son nom d’Eucalyptus globulus.
Aspect de la plante :
La famille des myrtacées, à laquelle appartient le genre Eucalyptus, a une origine
relativement récente. Comme à l’intérieur du genre les espèces ne sont pas encore
strictement fixées, elles se croisent souvent, étant donné la facilité avec laquelle les
graines de pollen se transfèrent d’une espèce à une autre. Cette extrême variabilité
se reflète dans certaines incertitudes ou complications de la nomenclature
systématique.
En tant que genre, l’Eucalyptus se reconnaît facilement mais les différentes espèces
se différencient au contraire difficilement. La beauté de l’Eucalyptus réside dans son
feuillage. En effet ses feuilles à géométrie variable, qui sont persistantes se modifient
avec astuce selon les exigences, durant leurs deux années de croissance. Elles
commencent par être rondes et étalées horizontalement sans pétioles, directement
rattachées aux rameaux par paires. Recouvertes d’une cire grisâtre, elles ont un
aspect argenté. Leur état juvénile demeure un certain temps chez les arbres jeunes
mais persiste chez les mâles nains. Ensuite passant par divers stades intermédiaires,
elles se font longues, étroites sans faces différées, puis se mettent à pendre
verticalement pour échapper à l’aplomb du soleil. A ce moment là, elles annoncent la
couleur de leurs espèces ; des teintes pouvant aller du vert pâle au bronze mat, du
gris clair au bleu le plus tendre. Chaque feuille contient un semis de mini-glandes
exsudant des sécrétions huileuses, ses huiles appelées essences sont très
odoriférantes.

5
Récolte :
L'Eucalyptus globulus ne supporte pas les températures au dessous de +4°C. Sa
multiplication se pratique par semis des graines à l'automne, dans des godets, sous
serre. Repiquez les plants au printemps. Récoltez les feuilles durant tout l'été, en
choisissant les plus allongées et étroites, portées par les branches plus âgées. Faites-
les sécher à l'ombre dans des locaux aérés
Actifs
Les feuilles de l'eucalyptus globulus renferment des tanins, de l'alcool cérylique, un
diphénol (pyrocatéchine), une résine acide et surtout 5 à 7% d'huile essentielle aux
composants multiples, le plus notable étant l'eucalyptol. C’est dans les feuilles âgées
de l’eucalyptus que l’on trouve la plus forte concentration en eucalyptol.
Utilisation des huiles essentielles d’Eucalyptus :
Compte tenu de la diversité de leur composition chimique que ce soit du point de vue
qualitatif ou quantitatif, les huiles essentielles de feuilles d’Eucalyptus trouvent leurs
emplois dans différents secteurs.
En pharmacie : Leur utilisation est attribuée à son constituant principal le 1,8 cinéole
(ou eucalyptol). Notons que la qualité médicinale de l’essence d’Eucalyptus doit obéir
aux pharmacopées internationales qui exigent une teneur minimale en eucalyptol de
70 % et une absence ou traces de phéllandrène. Outre le 1,8 cinéole qui se révèle être
un excellent antiseptique pulmonaire, les autre constituants de l’huile semblent
intervenir également en augmentant de façon notable son pouvoir bactéricide.
L’huile essentielle d’Eucalyptus qui est toxique à forte dose fait l’objet de prés de 200
préparations pharmaceutiques principalement comme désinfectant des voies
respiratoires, mais aussi comme vermifuge, cicatrisant etc … .
En parfumerie : C’est le débouché principal de la majorité des essences non
médicinales. En effet le citronellal, composé principal de l’huile d’E.citriodora est
utilisé directement comme parfum ou pour en produire d’autres.
 6
7
8
9
10
11
12
13
14
15
16
17
6
7
8
9
10
11
12
13
14
15
16
17
1
/
17
100%