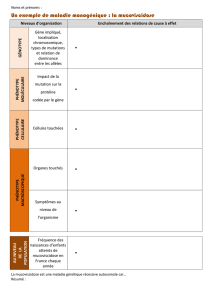
Dossier Atteinte digestive de la mucoviscidose chez l`enfant

Dossier
Atteinte digestive
de la mucoviscidose
chez l’enfant
Anne Munck
Hôpital Robert Debré 48, bd Serurier 75019 Paris
Résumé
Les troubles digestifs liés à l’atteinte pancréatique ou du tractus digestif sont
fréquents chez les enfants atteints de mucoviscidose. Établir le diagnostic
étiologique nécessite une démarche diagnostique rigoureuse car, en plus des
étiologies rencontrées chez les sujets sains, il faut rechercher celles spécifiques
à la maladie.
Il ne faut pas sous-estimer une pathologie abdominale qui pourrait passer au
deuxième plan chez ces patients pour lesquels la préoccupation médicale est
d’abord d’ordre pulmonaire.
Les troubles digestifs risquent – s’ils sont négligés – d’entraîner une
dénutrition qui à son tour influe de façon significative sur le pronostic global de
la maladie.
Les prises en charge digestive et nutritionnelle doivent faire partie intégrante de
la prise en charge médicale au même titre que les traitements à visée pulmo-
naire.
Mots clés : mucoviscidose, insuffisance pancréatique, douleur abdominale, symptomatologie
digestive
La symptomatologie digestive de la
mucoviscidose comporte des ma-
nifestations pancréatiques et gastro-
intestinales. Les propriétés d’absorp-
tion, de digestion et de motricité sont
modifiées tout au long du tractus di-
gestif. La démarche diagnostique s’ap-
puie prioritairement sur l’interroga-
toire et l’examen clinique afin de
limiter les examens complémentaires
chez les patients déjà très sollicités sur
le plan des investigations et des traite-
ments. Seules les manifestations di-
gestives spécifiques à la maladie se-
ront abordées dans cette mise au
point.
Manifestations
pancréatiques
Insuffisance pancréatique
exocrine
Manifestation classique de la mu-
coviscidose, l’insuffisance pancréati-
que exocrine (IPE) est la principale
cause des pertes énergétiques au
cours de la mucoviscidose. Elle est
symptomatique lorsque environ 98 %
de la fonction pancréatique a disparu
[1]. L’involution de la glande est très
précoce, parfois anténatale [2] mais
peut se compléter au fil de la vie des
patients qui peuvent être initialement
« suffisants » pancréatiques, d’où la
nécessité d’une surveillance régulière
m
t
p
Tirés à part : A. Munck
mt pédiatrie, vol. 8, n° 3, mai-juin 2005 197
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 04/06/2017.

de leur fonction pancréatique. Ainsi, les données de l’Ob-
servatoire national de la mucoviscidose [3] portant sur
3 400 patients identifient 87 % de malades sous extraits
pancréatiques.
Il existe une corrélation entre le génotype et le phéno-
type en ce qui concerne l’IPE : 97 % des patients homo-
zygotes pour la mutation DF508, 72 % des patients hété-
rozygotes composites pour la mutation DF508 et 36 % des
patients qui ne possèdent pas cette mutation [4] sont
insuffisants pancréatiques.
Le dysfonctionnement de la protéine CFTR est respon-
sable de l’hyperviscocité des sécrétions pancréatiques
appauvries en eau et en bicarbonates, qui obstruent les
canaux pancréatiques et contribuent à la destruction se-
condaire des acini. Le tissu acineux est progressivement
remplacé par du tissu fibreux, avant que n’apparaisse une
involution adipeuse. Les îlots de Langerhans sont quant à
eux longtemps épargnés.
La principale manifestation clinique de l’IPE est repré-
sentée par la stéatorrhée (émission de selles anormale-
ment graisseuses) souvent associée à des douleurs abdo-
minales, un ballonnement et un prolapsus rectal.
L’insuffisance de sécrétion en lipase, trypsine et chy-
motrypsine entraîne un syndrome de maldigestion et de
malabsorption des graisses et des protéines qui, malgré
l’augmentation de l’appétit, a un effet délétère sur l’état
nutritionnel avec un retard staturo-pondéral et des caren-
ces en vitamines liposolubles (A, D, E, K) et en acides gras
essentiels. Le déficit de la sécrétion en amylase pancréa-
tique est responsable d’une maldigestion de l’amidon,
aboutissant à une fermentation bactérienne colique ac-
crue dont les conséquences nutritionnelles sont mal
connues.
L’évaluation précise de la sécrétion enzymatique du
pancréas nécessiterait la réalisation d’un tubage duodé-
nal. Ce test invasif n’est jamais utilisé en pratique clini-
que : il est réservé à la réalisation de tests pharmacologi-
ques en recherche clinique. L’étude de la stéatorrhée
mesure la quantité de graisses fécales (en g/24 heures) au
cours du recueil de selles réalisé pendant 3 jours consé-
cutifs [5] ; elle est pathologique au-delà de 3,5 à 4 g/24 h
Même en cas d’IPE sévère, sa sensibilité ne dépasse pas
70 %, et sa spécificité est de l’ordre de 60 à 75 %. Chez
l’enfant, on utilise volontiers la mesure du coefficient
d’absorption des graisses :
(graisses ingérées −graisses excrétées)
(graisses ingérées) x 100
La stéatorrhée est considérée comme anormalement
élevée pour un coefficient d’absorption inférieur à 90-
93 % chez l’enfant de plus d’un an et de 85 % avant
6 mois. En dépit de ses limites et de ses inconvénients
(durée du recueil des selles), la mesure de la stéatorrhée
reste la principale méthode permettant d’évaluer objecti-
vement l’efficacité de la supplémentation en enzymes
pancréatiques.
La plupart des enzymes pancréatiques sont dégradées
au cours de leur transit intestinal. Le dosage des enzymes
résistantes comme l’élastase est considéré comme un as-
sez bon reflet de la sécrétion pancréatique exocrine ; ce
dosage est réalisé sur un seul échantillon de selles et les
valeurs normales se situent au-dessus de 200 lg/g de
selles : sa sensibilité est de l’ordre de 98 % pour les IPE
sévères et de 70 % pour les IPE modérées [6]. Son dosage
n’est pas influencé par la prise d’extraits pancréatiques.
Quand à la chymotrypsine fécale, sa sensibilité évaluée à
96 % lors d’IPE sévère tombe à 50 % pour les déficits
partiels ; la valeur du test peut être influencée par la prise
d’EP.
En pratique, le dosage de l’élastase fécale est de plus
en plus utilisé pour confirmer ou infirmer la présence
d’une IPE chez un enfant suspect ou atteint de mucovisci-
dose, alors que la mesure de la stéatorrhée a pour but de
contrôler, si besoin, l’efficacité du traitement substitutif et
d’adapter la posologie des extraits pancréatiques. Enfin,
une méthode d’avenir pourrait être le test respiratoire aux
lipides marqués [7].
Le traitement de l’insuffisance pancréatique exocrine
repose sur l’utilisation par voie orale des extraits pancréa-
tiques (EP). La mise sur le marché, il y a une quinzaine
d’années, de préparations enzymatiques avec un enro-
bage gastrorésistant (évitant leur inactivation par l’acidité
gastrique) a représenté un progrès décisif autorisant une
alimentation libre, normale, riche en graisses. Les prépa-
rations enzymatiques disponibles en France se présentent
sous la forme de gélules contenant près d’une centaine de
microsphères (tableau 1) ; elles sont données au début (et
au milieu) du repas ou de la collation dès lors qu’ils
Tableau 1.Extraits pancréatiques disponibles en France
Nom commercial Gastro-protégé Présentation Activité enzymatique (Unités Ph Eur)
Lipolytique Protéolytique Amylolytique
Créon
®
12000 Oui Gélule 12 000 700 12 000
Créon
®
25000 Oui Gélule 25 000 1 000 18 000
Eurobiol
®
25000 Oui Gélule 25 000 1250 2 2500
Kreon fûr Kinder Oui Mesure 5 000 200 3 600
Atteinte digestive de la mucoviscidose chez l’enfant
mt pédiatrie, vol. 8, n° 3, mai-juin 2005
198
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 04/06/2017.

contiennent des graisses [8]. Leur taille varie de 1,2 à
2 mm, et ce petit diamètre permet un passage transpylori-
que relativement synchrone du repas. Les microsphères ne
doivent pas être croquées.
Le but de la supplémentation est d’obtenir une absorp-
tion intestinale proche de la normale des graisses et des
protéines. En pratique clinique, on se contente le plus
souvent de vérifier la bonne réponse du patient au traite-
ment médical : absence de douleurs abdominales et/ou de
diarrhée graisseuse, croissance staturo-pondérale régu-
lière. L’augmentation excessive des doses d’EP expose à
l’apparition d’une colopathie fibrosante [9]. Il s’agit d’un
tableau non spécifique d’obstruction intestinale distale
résistant au traitement médical, parfois associé à une
diarrhée sanglante. Les cas décrits ont dû subir une inter-
vention chirurgicale qui objectivait une sténose du colon
ascendant nécessitant le plus souvent une hémicolecto-
mie droite ; l’examen anatomopathologique mettait en
évidence une fibrose sous-muqueuse importante, respon-
sable d’une sténose intraluminale du colon. Les facteurs
de risque d’apparition d’une colopathie fibrosante sont
l’utilisation dans les 12 mois précédant la chirurgie d’EP
fortement dosés avec une posologie quotidienne très éle-
vée et l’utilisation de laxatifs [10]. La physiopathologie des
lésions coliques reste encore mystérieuse. Les lésions ob-
servées pourraient être provoquées par un ou plusieurs des
constituants des EP ; une observation a cependant été
rapportée chez un bébé qui n’avait pas encore reçu d’EP
[11].
La conférence de consensus sur l’utilisation des EP,
organisée en mars 1995 sous l’égide de la Cystic Fibrosis
Foundation et de la Food and Drug Administration, n’a pas
remis en cause l’intérêt d’un régime hypercalorique, donc
relativement riche en graisses [12]. C’est l’efficacité des EP
gastroprotégés qui a permis d’augmenter l’apport en grais-
ses et donc de couvrir les besoins énergétiques majorés
des patients [13]. Chez le nourrisson, la dose recomman-
dée d’EP varie de 2 000 à 4 000 unités lipase/120 mL de
lait ou par tétée chez l’enfant au sein.
Pour plus de commodité, on peut également exprimer
les recommandations en EP par rapport au poids : la dose
initiale d’EP devrait être de 1 000 unités lipase/kg/repas
avant l’âge de 4 ans et 500 unités lipase/kg/repas après
l’âge de 4 ans. La dose est divisée par 2 pour les colla-
tions ; la dose quotidienne totale est estimée pour une
moyenne de 3 repas et 2 ou 3 collations par jour.
Lorsque les symptômes de malabsorption (diarrhée
graisseuse, douleurs abdominales, stéatorrhée excessive)
persistent, il faut rechercher des facteurs de mauvaise
efficacité des EP avant d’en augmenter la posologie : uti-
lisation de produits périmés ou soumis à une chaleur
excessive ; prise des EP en dehors et non pas au début des
repas ; consommation excessive de jus de fruits ; absence
de prise d’EP lors de la consommation de lait et/ou la prise
d’une collation ; mauvaise observance du patient (refus de
la maladie, souhait des adolescentes de rester mince).
L’augmentation des doses d’EP n’est décidée qu’après
avis médical. Au-delà d’une dose d’EP de 2 500 unités
lipase/kg/repas, la recherche d’une pathologie digestive
associée à la mucoviscidose s’impose : maladie cœliaque,
intolérance au lactose, pullulation microbienne intesti-
nale, infection bactérienne ou parasitaire, maladie de
Crohn. Il est recommandé de ne pas dépasser la dose de
10 000 unités lipase/kg/j, avec un maximum de 250 000
unités lipase/j [14]. Actuellement la recherche s’oriente
vers le développement de lipases résistantes en pH acide,
notamment la lipase gastrique d’origine canine, produite
par génie génétique, qui pourrait être dans l’avenir asso-
ciée aux enzymes pancréatiques gastroprotégées.
Lorsque la stéatorrhée reste excessive, la prescription
concomitante d’un adjuvant thérapeutique, destiné à ré-
duire l’hypersécrétion gastrique acide qui peut gêner l’ac-
tivité des enzymes pancréatiques, devient justifiée : les
inhibiteurs de la pompe à protons en une prise quoti-
dienne permettent une amélioration significative, de l’or-
dre de 20 % par rapport aux valeurs sous EP seuls, de
l’excrétion fécale des graisses [15].
Pancréatite aiguë
Au cours des formes atténuées de mucoviscidose (il
existe une très forte corrélation entre pancréatite aiguë et
génotype CFTR modéré), la fonction pancréatique exo-
crine est préservée mais le dysfonctionnement des canaux
chlore favorise la précipitation des secrétions acineuses
entraînant des poussées de pancréatites récurrentes chez
près de 10 à 15 % des patients suffisants pancréatiques
[16]. Des complications pancréatique locales ou systémi-
ques sont rarement décrites [17]. La pancréatite aiguë peut
être le premier élément clinique aboutissant au diagnostic
de mucoviscidose chez l’adulte notamment [17] mais
rarement chez l’enfant [18] ; le test de la sueur et la
recherche des mutations du gène CFTR doivent faire partie
du bilan étiologique d’une pancréatite aiguë.
Manifestations gastro-intestinales
Le reflux gastro-œsophagien
La fréquence du reflux gastro-œsophagien (RGO) est
élevée dans la mucoviscidose comme cela a pu être
évalué par les études pH-métriques [19]. Des brûlures
épigastriques ou des régurgitations très évocatrices de
reflux sont mentionnées par plus de 20 % des patients. À
l’âge pédiatrique, sa fréquence est encore plus importante,
jusqu’à 76 % selon Vic et al. [20]. On ne sait pas préciser
la fréquence avec laquelle le reflux aggrave la maladie
pulmonaire ou vice-versa, mais il y a vraisemblablement
une interaction entre ces deux pathologies, qui contribue
d’ailleurs à la malnutrition, puisque le RGO peut majorer
les pertes (régurgitations) et diminuer les ingesta (dyspha-
mt pédiatrie, vol. 8, n° 3, mai-juin 2005 199
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 04/06/2017.

gie), et la maladie respiratoire peut majorer les besoins
caloriques (travail ventilatoire) et diminuer les ingesta
(anorexie).
La pathogénie du RGO est multifactorielle : les relaxa-
tions inappropriées du sphincter inférieur de l’œsophage
(SIO) [21] mais également l’élévation de la pression
thoraco-abdominale, les expirations prolongées de la ki-
nésithérapie [22], les médicaments alpha-adrénergiques
et la théophylline. Une hypersécrétion gastrique est sou-
vent signalée chez les patients, rendant le RGO plus
agressif pour la muqueuse œsophagienne.
À son tour, le RGO pourrait de façon insidieuse majo-
rer la symptomatologie respiratoire, soit par des micro-
aspirations, soit par l’œsophagite qui altère des phénomè-
nes réflexes naissant dans l’œsophage [23]. Ces éléments
poussent à dépister précocement le RGO et à le traiter. La
présentation du RGO n’a pas de spécificité clinique mais
nécessite une prise en charge efficace ; il peut s’agir de
régurgitations, de malaises, de brûlures rétrosternales, de
douleurs épigastriques, de dysphagie, d’hématémèse et de
signes respiratoires qui répondent mal à la thérapeutique
habituelle. Une baisse des ingesta avec une préférence
pour les aliments liquides ou une dysphagie font craindre
une sténose œsophagienne.
Les explorations peuvent comporter un transit baryté
œsogastroduodénal qui vérifiera l’absence d’anomalie
anatomique telle qu’une malrotation ou un pancréas an-
nulaire. Il permet de visualiser un reflux, une hernie hia-
tale, et une sténose de l’œsophage. Devant des brûlures
rétrosternales, une douleur épigastrique ou une hématé-
mèse, la fibroscopie œsogastroduodénale (FODG) doit
être réalisée. La pH-métrie de longue durée reste l’examen
paraclinique le plus sensible et le plus spécifique lors de
diagnostic douteux de RGO.
La prise en charge thérapeutique est débutée dès
l’identification du reflux, qu’il soit peu symptomatique ou
cliniquement parlant. Les prokinétiques utilisés sont le
métoclopramide ou le domperidium (le cisapride n’est
plus autorisé en France). En cas de reflux sévère avec
œsophagite, il faut réduire la sécrétion d’acide gastrique
par des anti-H
2
ou des inhibiteurs de la pompe à protons.
Lorsque, malgré un traitement médical bien conduit, le
RGO persiste et qu’une gastrostomie est envisagée ou
lorsque le RGO se complique de perte pondérale, d’œso-
phagite érosive, de majoration des symptômes respiratoi-
res ou de sténose de l’œsophage, il faut envisager une
fundoplicature de Nissen ; cette intervention doit être
réalisée dans des centres de référence pour la mucovisci-
dose.
Constipation
La constipation est un problème pour bon nombre de
ces patients. Il faut la distinguer du SOID et éliminer les
causes de constipation retrouvées également dans la po-
pulation générale. Le transit intestinal doit être apprécié à
chaque visite, parallèlement à la palpation abdominale.
On peut retrouver des fissures anales. Il est utile d’enrichir
le régime en fibres et d’augmenter l’apport hydrique.
L’abord thérapeutique de la constipation est identique à
celui des autres patients indemnes de mucoviscidose,
avec de plus la possibilité d’utiliser des agents mucolyti-
ques (N-acétylcystéine ou des solutions de type polyéthy-
lène glycol) par voie orale. La constipation ne doit en
aucun cas être traitée par une réduction de posologie des
extraits pancréatiques [24].
Syndromes d’obstruction intestinale distale
Le syndrome d’obstruction intestinale distale (SOID),
ou équivalent d’iléus méconial, est une pathologie récur-
rente parfois chronique, spécifique à la mucoviscidose. Il
est dû à une obstruction de sévérité variable de l’intestin
grêle, débutant généralement dans la région iléocæcale ;
le tube digestif est épaissi, œdématié et rempli de matériel
compact [25].
Le patient se plaint de douleurs abdominales à type de
crampes, souvent dans la fosse iliaque droite, associées à
une distension abdominale, une anorexie et une perte de
poids variable. La palpation peut retrouver une masse de
la fosse iliaque droite comme dans les mucocèles appen-
diculaires. Le transit peut être conservé et peu modifié par
rapport aux habitudes du patient. L’obstruction intestinale
est le plus souvent partielle mais peut devenir totale avec
des vomissements et une distension abdominale majeure.
Cette symptomatologie peut être révélatrice d’une muco-
viscidose et incite à réaliser un test de la sueur.
Il est difficile d’apprécier la fréquence du SOID étant
donné la grande variabilité des symptômes ; il semble que
15 % des patients en souffrent, quels que soient le sexe et
l’âge.
L’étiologie du SOID reste méconnue ; il survient à de
rares exceptions près chez des patients insuffisants pan-
créatiques. Les facteurs pouvant jouer un rôle dans le
déclenchement du SOID sont les anomalies des mucines
intestinales, le défaut de transport des électrolytes, l’allon-
gement du temps de transit orocæcal. On n’identifie pas
de facteur déclenchant univoque, mais une relative dés-
hydratation, des changements diététiques ou une supplé-
mentation inappropriée en extraits pancréatiques sem-
blent intervenir.
Établir le diagnostic étiologique des douleurs abdomi-
nales dans la mucoviscidose peut être très délicat (ta-
bleau 2). Il peut s’agir d’appendicite dont le diagnostic est
souvent difficile car on peut avoir une simple distension de
l’appendice par du mucus, parfois un tableau typique
d’appendicite aiguë mais également des complications de
type occlusion, abcès ou perforation, favorisées d’une part
par la prescription fréquente d’antibiotiques qui abâtardit
le tableau clinique, et d’autre part par la fréquence des
douleurs abdominales au cours de la mucoviscidose, à tort
banalisées.
Atteinte digestive de la mucoviscidose chez l’enfant
mt pédiatrie, vol. 8, n° 3, mai-juin 2005
200
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 04/06/2017.

L’invagination intestinale peut être chronique et répé-
tée dans la mucoviscidose, notamment chez le grand
enfant, et la présentation clinique est souvent modérée
sans présence de sang dans les selles [26].
Un volvulus d’une anse grêle peut survenir et compli-
quer un SOID (ou indépendamment de celui-ci) ; l’ur-
gence diagnostique est réelle.
Le diagnostic de SOID est posé cliniquement devant
des symptômes d’obstruction intestinale tels qu’un arrêt
du transit, des vomissements répétés, voire bilieux, et
d’éventuels signes d’irritation péritonéale, dans un tableau
de douleurs abdominales à type de crampes localisées à la
fosse iliaque droite. La radiographie d’ASP, debout de
face, montre le degré d’obstruction et la présence de selles
granitées dans la fosse iliaque droite avec quelques ni-
veaux hydroaériques et une certaine distension sus-
jacente. En cas de doute diagnostique, le bilan est com-
plété par un examen cytobactériologique des urines, un
bilan hépatique complet, un dosage de l’amylasémie et de
la lipasémie, une opacification digestive (un lavement aux
macromolécules hydrosolubles hyperosmolaires peut
aider au diagnostic et représente un traitement efficace),
une échographie abdomino-pelvienne, voire un scanner
abdominal.
Une fois le diagnostic établi avec certitude, après avoir
éliminé une appendicite ou une occlusion complète, on
prescrit une solution de polyéthylène glycol par voie orale
à une posologie de 20-40 mL/kg/h ; le résultat de la vi-
dange est obtenu au bout de2à6h.Latolérance peut être
améliorée avec la prescription d’un prokinétique car on
observe souvent des nausées et un ballonnement [27]. Le
lavement aux macromolécules hydrosolubles hyperosmo-
laires est une alternative. Il faut se méfier de l’apparition de
troubles hydroélectrolytiques, l’opacification de la der-
nière anse iléale peut nécessiter des volumes importants.
La résolution complète des symptômes doit être obte-
nue et la radiographie d’ASP peut être utilisée pour mon-
trer la disparition du SOID. En cas de formes débutantes de
SOID, l’utilisation de N-acétylcystéine peut aider à la
résolution des symptômes.
Une forme compliquée d’occlusion nécessite une in-
tervention chirurgicale après mise en condition du patient
(l’utilisation de polyéthylène-glycol est alors formellement
contre-indiquée).
Étant donné le risque de récidive des épisodes, il est
important de préciser quelques mesures thérapeutiques
préventives, telles l’utilisation de médicaments prokinéti-
ques, et l’optimisation des médicaments qui améliorent la
digestion et l’absorption des graisses.
Pathologie appendiculaire
•Appendicite aiguë. La survenue d’une appendicite
serait plus rare (1-2 %) que dans la population générale
(7 %) [28] ; le diagnostic peut être masqué ou faussement
suggéré par d’autres pathologies (SOID) et les signes cli-
niques peuvent être d’interprétation difficile si la sympto-
matologie est abâtardie par une antibiothérapie concomi-
tante [29]. La fréquence des formes compliquées d’abcès
ou d’IIA est soulignée par certains auteurs. L’appendicite
est à distinguer d’une entité spécifique à la mucovisci-
dose : le mucocèle appendiculaire.
•Mucocèle appendiculaire. Evoqué devant l’associa-
tion de douleurs abdominales répétitives avec une masse
oblongue palpée dans la fosse iliaque droite sans symptô-
mes inflammatoires, il est repérable par l’échographie
abdominale centrée sur la FID (appendice épais distendu
par le mucus épais) et éventuellement confirmée par
l’opacification (lavement baryté). L’intervention consiste
en la réalisation d’une appendicectomie avec ablation
d’une collerette cæcale pour éviter les récidives [30] ; elle
est indiquée lors de symptomatologie douloureuse afin
d’éviter les formes compliquées d’abcès ou de perforation
en amont.
Prolapsus rectal
Plus fréquent au moment de l’apprentissage de la
propreté, on peut cependant l’observer quel que soit l’âge.
La constipation, la diarrhée et la malnutrition sont des
facteurs favorisants, de même que des efforts de toux. Un
régime appauvri en graisses ne permet pas de le prévenir.
Une meilleure adaptation de la posologie des extraits
pancréatiques et une éducation sphinctérienne de la défé-
cation, contribuent à la réduction des épisodes de prolap-
sus. Une indication chirurgicale peut se discuter de façon
tout à fait exceptionnelle lors de douleurs importantes, ou
lorsqu’une incontinence apparaît à chaque épisode de
prolapsus [31].
L’iléus méconial
L’iléus méconial est une occlusion intestinale néona-
tale liée à l’absence de progression du méconium au
niveau de l’iléon terminal. Cela représente le symptôme
clinique le plus précoce de mucoviscidose, révélateur
chez 10 à 20 % des patients. La majorité des nourrissons
présentant un iléus méconial a une mucoviscidose. Cette
présentation clinique peut survenir quelles que soient les
mutations du nourrisson ; elle n’est pas l’apanage exclusif
des cas avec insuffisance pancréatique exocrine (IPE),
même si l’IPE est presque toujours présente.
Tableau 2.Diagnostic différentiel du syndrome d’obstruction
intestinale distale (SOID)
Appendicite et ses complications, mucocèle appendiculaire
Invagination intestinale
Volvulus
Brides post-opératoires
Pathologie tubo-ovarienne
Cholélithiase
Pancréatite
Ulcère peptique
mt pédiatrie, vol. 8, n° 3, mai-juin 2005 201
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 04/06/2017.
 6
7
6
7
1
/
7
100%