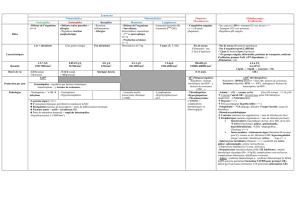
Graphique 1

UNIVERSITESIDIMOHAMMEDBENABDELLAH
FACULTEDEMEDECINEETDEPHARMACIE
FES
AnnéeThèseN°/201008110
UNIVERSITESIDIMOHAMMED
BENABDELLAH
FES
L’APLASIEMEDULLAIRECHEZL’ENFANT
(Etudede08casetrevuedelalittérature)
THESE
PRESENTEEETSOUTENUEPUBLIQUEMENTLE26/04/2010
PAR
Néele17Avril1984àBerkane
Mlle.BAALISANAE
POURL'OBTENTIONDUDOCTORATENMEDECINE
MOTS-CLES:
Aplasiemédullaire-Enfant-AnémiedeFanconi-Greffedemoelle
JURY
M. Professeur
M. Professeur
.Professeur
Mme.BONOWAFAA
Professeur
HIDAMOUSTAPHA........................................................
dePédiatrie .............................................
de
M.............................................................
agrégéde
...........................................................
agrégédeMédecineinterne
BOUHARROUABDELHAK
Pédiatrie
ATMANISAMIR Pédiatrie JUGE
PRESIDENTETRAPPORTEUR

~ 1 ~
Sommaire

~ 2 ~
1. Introduction.................................................................................................... 4
2. Rappel physiopathologique ............................................................................. 7
3. Matériel et méthodes d'études: ...................................................................... 10
4. Résultats: ...................................................................................................... 46
► Epidémiologie ......................................................................................... 47
► Clinique .................................................................................................. 50
► Para clinique ........................................................................................... 52
► Etiologies ............................................................................................... 54
► Traitement ............................................................................................. 55
► Evolution ................................................................................................ 55
5. Discussion: .................................................................................................... 56
► Epidémiologie ......................................................................................... 58
► Clinique ................................................................................................. 59
►Para clinique: ........................................................................................... 61
◦NFS ....................................................................................................... 61
◦Myélogramme ....................................................................................... 62
◦BOM ..................................................................................................... 63
◦Autres .................................................................................................. 68
► Etiologies: ........................................................................................ 68
◦Constitutionnelles ................................................................................. 68
◦Acquises ............................................................................................... 80
◦ Idiopathiques ....................................................................................... 86
►Traitement .............................................................................................. 86
6. Evolution et Pronostic ................................................................................... 101
7. Suivi ......................................................................................................... 105
8. Conclusion ................................................................................................... 108
9. Résumés ....................................................................................................... 111
10. Bibliographie .............................................................................................. 116

~ 3 ~
LEXIQUE DES ABREVIATIONS
AM: Aplasie médullaire
CSH: Cellules souches hématopoïétiques
MH: Motif d'hospitalisation
ATCD: Antécédents
HDM: Histoire de la maladie
FR: fréquence respiratoire
FC: fréquence cardiaque
TA: Tension artérielle
DS: Déviations Standards
RSP: Retard staturo-pondéral
NFS: Numération formule Sanguine
Hb: hémoglobine
VGM: volume globulaire moyen
CCMH: Concentration corpusculaire moyenne en hémoglobine
GB: Globules blancs
Plq: Plaquettes
PNN: Polynucléaires neutrophiles
TP: Temps de Prothrombine
TCK: Temps de Cephaline Kaolin
BOM: biopsie ostéomédullaire
Sd: syndrome
PEC: prise en charge
C3G: Céphalosporines 3ème génération

~ 4 ~
INTRODUCTION
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
1
/
129
100%