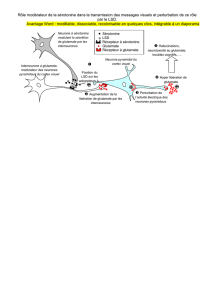
Lire l`article complet

La Lettre du Neurologue • Vol. XIV - n° 11 - décembre 2010 | 407
DOSSIER THÉMATIQUE
Voie glutamatergique
et maladie d’Alzheimer
P
rincipal neurotransmetteur excitateur du
cerveau, le glutamate joue un rôle majeur
dans le fonctionnement cérébral, depuis
les premières étapes de la neurogenèse jusqu’au
vieillissement cérébral. Son degré d’implication
dans de nombreux processus physiologiques est
corroboré par la responsabilité qu’il endosse dans
la genèse de nombreux processus pathologiques. En
effet, on a pu mettre en évidence un dysfonction-
nement de la transmission glutamatergique dans de
nombreuses maladies neurologiques telles que l’épi-
lepsie, l’accident vasculaire cérébral, et la plupart des
maladies neurodégénératives comme les démences,
la sclérose latérale amyotrophique ou la maladie de
Parkinson (1). En outre, l’approche pharmacologique
des troubles psychiatriques a permis d’établir un lien
entre le glutamate et certaines maladies mentales,
notamment la schizophrénie (2). La multiplicité de
ses sites d’action rend difficile la réalisation d’une
revue exhaustive sur les fonctions cérébrales du
glutamate. Aussi, nous avons voulu, au travers de
cet article, illustrer certains processus centraux où le
glutamate joue un rôle clé et évoquer le lien qui existe
entre le fonctionnement cérébral normal et la patho-
logie. Après un bref rappel de l’organisation générale
du système glutamatergique dans le cerveau, nous
examinerons la place du glutamate dans le neuro-
développement, la motricité, les fonctions cognitives
et les pathologies psychiatriques.
Organisation du système
glutamatergique
Le glutamate est le principal neurotransmetteur
excitateur du cerveau du mammifère, où il est
retrouvé dans 40% des synapses (1). Il constitue
le neurotransmetteur des cellules pyramidales
qui représentent la source des voies efférentes du
cortex. On le trouve dans les grandes voies neuro-
nales cortico-sous-corticales et, en particulier, dans
celles projetant vers les ganglions de la base, dans
les voies associatives cortico-corticales inter- et
intra-hémisphériques notamment à destination du
cortex limbique et dans l’hippocampe (3). Il agit sur
trois types de récepteurs postsynaptiques ionotro-
piques permettant l’ouverture d’un canal cationique :
NMDA (N-méthyl-D-aspartate), AMPA (α-amino-
3-hydroxy-5-méthyl-4-isoxazole propionate) et
kaïnate. En outre, le glutamate se fixe également
sur trois types de récepteurs métabotropiques,
localisés en pré- ou postsynaptiques, qui activent la
phospholipase C (entraînant la production d’inositol
triphosphate et de diacylglycérol) ou inhibent l’adé-
nylate cyclase (diminuant la production d’AMPc).
L’ensemble de ces récepteurs est localisé sur les
neurones mais également sur les cellules gliales (4).
Glutamate
et neurodéveloppement
Au cours de la neurogenèse, la maturation et la
migration neuronales s’opèrent de façon concomi-
tante. Il ressort d’études sur l’hippocampe de rat que
la maturation des synapses GABAergiques survient
en premier et que le GABA reste l’unique neurotrans-
metteur capable de générer des activités synaptiques
jusqu’aux derniers jours de la vie embryonnaire. En
outre, en raison du degré de maturation des cotrans-
porteurs K-Cl, la concentration intracellulaire de
Cl
–
est élevée durant la vie embryonnaire et jusqu’à
la deuxième semaine postnatale. Aussi, à ce stade,
le GABA présente un rôle dépolarisant nécessaire à
la mise en place du réseau, dont les seules activités
détectables sont constituées par la récurrence à
Glutamate
et grandes fonctions cérébrales
Glutamate and brain functions
A. Eusebio*, **, J. Micallef-Roll*
* Centre d’investigation clinique
CPCET, hôpital de la Timone, CHU de
Marseille.
** Service de neurologie et de
patho logie du mouvement, pôle de
neurosciences cliniques, hôpital de
la Timone, CHU de Marseille.

408 | La Lettre du Neurologue • Vol. XIV - n° 11 - décembre 2010
Résumé
0,1 Hz de potentiels géants (5). L’apparition de
synapses glutamatergiques matures, exprimant
d’abord le récepteur NMDA puis AMPA, coïncide
avec l’inversion du gradient chlore et l’apparition de
l’activité hyperpolarisante du GABA. Ainsi, l’absence
du glutamate durant les premières étapes de la
neurogenèse est compensée par le rôle dépolarisant
transitoire du GABA.
Toutefois, les récepteurs du GABA et du glutamate
sont exprimés par les neurones immatures avant la
formation des synapses (6). C’est par ce biais que
ces neurotransmetteurs exercent un rôle dans la
migration des neurones immatures. Le GABA et
l’activation des récepteurs NMDA exercent un rôle
crucial dans la migration des neurones glutama-
tergiques. Au cours de leur migration, les interneu-
rones libèrent du GABA qui module la migration
des neurones glutamatergiques (6). À l’inverse,
il a récemment été montré que la migration des
interneurones est essentiellement modulée par
l’activation des récepteurs AMPA (7). Les synapses
glutamatergiques ne deviennent matures que
lorsque les interneurones cibles ont atteint un degré
de maturation suffisant (5).
Glutamate et contrôle moteur
Le contrôle moteur est assuré essentiellement par
le cervelet et les ganglions de la base. Ces derniers
sont constitués par un ensemble de noyaux sous-
corticaux organisés en réseau ; ils jouent un rôle
dans la motricité et sont en outre impliqués dans les
processus d’apprentissage. Le striatum représente la
structure d’entrée principale de ce réseau en recevant
des afférences glutamatergiques d’origine corticale
et thalamique. Il reçoit également des afférences
dopaminergiques en provenance de la substance
noire pars compacta. Les noyaux de sortie du réseau
sont représentés par le globus pallidus interne et
la substance noire pars reticulata qui, tous deux,
émettent des projections GABAergiques inhibitrices
en direction du thalamus qui projette à son tour vers
le cortex (8). La modulation du mouvement est donc
sous la dépendance d’une boucle GABA-glutamate
modulée par la dopamine. En effet, cette dernière
exerce une action inhibitrice sur la transmission
glutamatergique cortico-striale par l’intermédiaire
de récepteurs D2 localisés sur le versant présynap-
tique de la synapse glutamatergique (9). Cette action
permet de favoriser l’activation de certains neurones
striataux et agit comme un filtre de l’information
glutamatergique corticale (9).
L’étude de modèles animaux de maladies neuro-
dégénératives a permis de préciser l’interaction
existant entre la dopamine et le glutamate. Ainsi,
de nombreuses études in vitro ont montré l’exis-
tence d’une hyperactivité glutamatergique striatale
dans le modèle du rat parkinsonien, obtenu par
injection de 6-hydroxydopamine (6-OHDA) dans
la substance noire pars compacta (10). Il a ensuite
été montré que le traitement des animaux parkinso-
niens avec la L-dopa permettait de supprimer cette
hyperactivité glutamatergique, suggérant que les
effets bénéfiques moteurs de ce traitement sont liés
à la réduction de l’hyperactivité glutamatergique
(11). De même, la lésion du noyau subthalamique,
utilisée en traitement de la maladie de Parkinson
avant l’avènement des techniques de stimulation
cérébrale profonde, entraîne la suppression de cette
hyperactivité dans ce même modèle de rat parkin-
sonien (12). Par ailleurs, c’est en étudiant ces modèles
animaux que le rôle de la dopamine dans la plasticité
de la synapse glutamatergique a été mis en évidence.
En effet, la dénervation dopaminergique supprime
les phénomènes de potentialisation et de dépression
à long terme (PLT et DLT) [13]. Dans le cadre de la
synapse cortico-striatale, le phénomène de PLT est
considéré comme un mécanisme d’apprentissage
moteur et la dépotentialisation de cette PLT, induite
par l’application d’une stimulation à basse fréquence
peu après l’induction de la PLT, comme un mécanisme
de suppression de l’information motrice inutile (13).
En effet, il a été constaté que les dyskinésies dopa-
induites observées dans ce modèle animal sont
associées à la perte des capacités de dépotentiali-
sation des neurones striataux (14). Ces dyskinésies
sont également corrélées à l’augmentation de
l’activité glutamatergique dans ces neurones et à
l’apparition d’une composante médiée par les récep-
teurs NMDA (15). Il a d’ailleurs été montré qu’elles
diminuent lors de l’administration d’un antagoniste
des récepteurs NMDA (LY235959) dans un autre
modèle animal (singe MPTP) mais également chez
Le rôle clé du glutamate, principal neurotransmetteur excitateur du cerveau mammifère, dans les principales
étapes du fonctionnement cérébral, est connu depuis de nombreuses années. Toutefois, la mise en évidence
de son implication dans les processus physiopathologiques de nombreuses affections neuropsychiatriques
dégénératives ou non suggère une place bien plus importante dans des domaines tels que la motricité
et la cognition. Cet article a pour objectif de fournir quelques exemples de l’impact des perturbations
de la transmission glutamatergique sur le fonctionnement cérébral.
Mots-clés
Glutamate
Cognition
Motricité
Parkinson
Développement

La Lettre du Neurologue • Vol. XIV - n° 11 - décembre 2010 | 409
l’homme avec un autre antagoniste des récepteurs
NMDA (l’amantadine) [16]. L’utilisation d’un antago-
niste des récepteurs AMPA (NBQX) semble produire le
même type d’effets sur les dyskinésies (16). Toutefois,
en dehors des effets sur les dyskinésies dopa-induites,
ni la modulation pharmacologique de l’activité des
récepteurs NMDA ni celle des récepteurs AMPA ne
semblent apporter de bénéfice sur les symptômes
de la maladie de Parkinson à proprement parler. À
l’inverse, il semble que la modulation exercée par les
récepteurs métabotropiques du glutamate, jusqu’alors
peu étudiée, joue un rôle critique dans la symptoma-
tologie déficitaire de la maladie. En effet, l’adminis-
tration d’agonistes des récepteurs métabotropiques
du groupe II (LY379268 ou DCG-IV) ou d’antagonistes
des récepteurs du groupe I (MPEP) chez le rat réduit
l’akinésie, induite par l’injection de 6-OHDA, en
diminuant l’hyperactivité glutamatergique cortico-
striatale et celle du noyau subthalamique (17).
Le troisième larron de ce ménage à trois est l’acétyl-
choline. Libérée par les interneurones toniquement
activés (TAN) du striatum, elle favorise la trans-
mission synaptique NMDA par l’intermédiaire de
récepteurs muscariniques M1 et joue un rôle crucial
dans le phénomène de PLT (18).
En conclusion, dans le réseau des ganglions de la
base, le glutamate interagit de façon rapprochée
avec la dopamine et l’acétylcholine, qui modulent la
transmission de l’information motrice en provenance
du cortex. La perte de cette modulation au cours
d’une maladie de la motricité telle que la maladie de
Parkinson induit des perturbations profondes dans
ce système, caractérisées par une levée du filtre de
l’information glutamatergique.
Glutamate et cognition
Concernant les fonctions cognitives, le glutamate
est impliqué principalement dans les processus
d’apprentissage et de mémorisation. Les mécanismes
synaptiques sous-tendant ces processus sont repré-
sentés par les phénomènes de PLT et DLT (1). En
effet, le blocage des récepteurs de type NMDA par
l’acide D-2-amino-5-phosphonopentanoïque (AP-5)
bloque la PLT mais inhibe également la mémoire
spatiale, hautement sensible à une atteinte hippo-
campique, sans toutefois affecter la mémoire
visuelle (19). L’effet observé implique l’altération
sélective de mécanismes de mémoire associative
puisqu’il épargne les performances motrices, la
perception sensitive et les aspects motivationnels
de la tâche.
Les processus de mémorisation comportent plusieurs
étapes : l’encodage, la consolidation, le stockage
et la récupération de l’information. Des études de
pharmacologie comportementale chez l’animal
ont permis de déterminer lesquelles de ces étapes
étaient sous la dépendance du glutamate. Ainsi,
il semble que l’activation des récepteurs NMDA
soit essentiellement impliquée dans les processus
d’encodage et de consolidation, alors que les récep-
teurs de type AMPA jouent également un rôle dans
les processus de récupération (20). En effet, M. Day
et al. ont montré que, dans une tâche d’association
d’odeurs avec des endroits précis impliquant la
mémoire épisodique, les rats recevant un antago-
niste NMDA (AP-5) ne pouvaient pas encoder de
nouvelles associations, mais étaient capables de
retrouver les associations déjà mémorisées, alors
que ceux recevant un antagoniste AMPA (6-cyano-
7-nitroquinoxaline-2,3-dione [CNQX]) n’encodaient
aucune nouvelle association et ne retrouvaient pas
non plus les associations déjà mémorisées (21). Les
phénomènes de PLT et de DLT interviennent tous
deux, de façon parfois antagoniste, dans les diffé-
rentes étapes de la mémorisation (22).
Les processus de reconnaissance visuelle, qui
sont particulièrement atteints dans la maladie
d’Alzheimer, sont générés dans une région mésiale
du lobe temporal, différente de l’hippocampe :
le cortex périrhinal (20, 23). Ces processus sont
également sous la dépendance étroite du glutamate
via les récepteurs AMPA et NMDA (23). Le blocage
des récepteurs NMDA (par l’AP-5) avant l’encodage
entraîne une diminution de la mémoire de reconnais-
sance visuelle à long terme mais pas à court terme,
suggérant que ces récepteurs interviennent, dans le
cortex périrhinal, essentiellement dans les phases
de consolidation. En outre, le blocage des récep-
teurs NMDA juste avant le test de réintroduction
de l’objet encodé n’influe pas sur les capacités de
reconnaissance, alors que le blocage des récepteurs
AMPA (par le CNQX) perturbe la reconnaissance, ce
qui suggère que ces derniers sont surtout impliqués
dans les processus de récupération (23).
Mémantine
et effets cognitifs
La modulation par la mémantine de la transmission gluta-
matergique au niveau des régions hippocampiques, les
premières touchées par le processus de neurodégéné-
rescence de la maladie d’Alzheimer, expliquerait que la
mémantine améliore les capacités cognitives, en particulier
mnésiques, ou ralentisse leur dégradation.
R. Bordet

410 | La Lettre du Neurologue • Vol. XIV - n° 11 - décembre 2010
Glutamate et grandes fonctions cérébrales
DOSSIER THÉMATIQUE
Voie glutamatergique
et maladie d’Alzheimer
Ces données donnent à penser qu’il existe une
certaine homogénéité de l’implication différentielle
des différents types de récepteurs glutamatergiques
dans les différents processus de mémorisation et
ce, quels que soient le type de mémoire (visuelle,
épisodique) et la structure cérébrale impliquée
(hippocampe, cortex périrhinal) [19-23].
Au cours de la maladie d’Alzheimer, l’activation des
récepteurs NMDA suivant un mode tonique plutôt
que phasique pourrait être à l’origine à la fois des
lésions d’excitotoxicité et des perturbations des
processus de mémorisation (24).
Néanmoins, des données récentes suggèrent que
le glutamate est également impliqué dans d’autres
fonctions cognitives, notamment les fonctions
exécutives. La mémoire de travail est caractérisée
par le maintien d’une information durant un temps
très court, notamment au cours de l’exécution d’une
tâche, cette information conditionnant un compor-
tement ultérieur. Elle implique, entre autres struc-
tures, le cortex préfrontal (20). L’administration de
kétamine (un antagoniste des récepteurs NMDA) à
des doses ne provoquant pas d’effet dissociatif chez
le sujet sain entraîne néanmoins une altération de
certaines capacités de la mémoire de travail (25).
De même, les capacités de maintien et d’adaptation
d’une stratégie, également sous la dépendance
du lobe préfrontal, sont diminuées chez l’animal,
comme l’indique l’apparition de persévérations après
administration de l’antagoniste NMDA non compé-
titif MK801 (20, 26). Chez l’homme, ces capacités
sont évaluées cliniquement par le test de Wisconsin
et diminuent après administration de kétamine à
doses subanesthésiques chez le sujet sain (20, 27).
Enfin, l’activation de récepteurs de type NMDA est
aussi impliquée, en association avec les récepteurs
dopaminergiques de type D1 du réseau cortico-strial,
dans les mécanismes d’apprentissage avec prédiction
de récompense (28). Cette intégration de signaux
glutamatergique et dopaminergique constitue la
base du lien existant entre les processus de plasticité
à l’origine des mécanismes d’apprentissage et
ceux à l’origine des comportements adaptatifs et
addictifs (29).
En conclusion, le glutamate intervient à tous les
niveaux des processus de mémorisation et d’appren-
tissage par l’intermédiaire des récepteurs NMDA,
essentiellement pour les phases d’encodage et de
consolidation, et par le biais des récepteurs AMPA
pour les phases de récupération de l’information.
Glutamate et psychiatrie
Une littérature abondante est consacrée aux liens
existant entre le glutamate et certaines maladies
psychiatriques, notamment la schizophrénie. Outre
les symptômes positifs et négatifs, l’accent a été mis
ces dernières années sur la désorganisation concep-
tuelle et les troubles cognitifs et affectifs associés
à la pathologie schizophrénique. Il a été en effet
clairement montré que la sévérité des symptômes
négatifs et des altérations cognitives était un déter-
minant important du devenir des patients, en parti-
culier de la possibilité de reprendre des études ou
une activité professionnelle (30).
En effet, certaines fonctions cognitives sont néces-
saires pour l’apprentissage et la mémorisation de
capacités permettant d’avoir un fonctionnement
communautaire normal. Ces troubles cognitifs
s’installent précocement dans la schizophrénie
et concernent plusieurs secteurs des fonctions
supérieures (30). Les capacités attentionnelles
sont les premières à être perturbées. Les fonctions
exécutives sont atteintes plus tardivement, et
leur atteinte entraîne des difficultés à élaborer
une stratégie et un défaut de plasticité mentale.
La mémoire de travail et les capacités d’appren-
tissage sont également perturbées. Le modèle
neuro biologique admis pour rendre compte de la
physio pathologie de la schizophrénie est représenté
par une hyperdopaminergie mésolimbique sous-
tendant les symptômes positifs (idées délirantes) et
une hypodopaminergie mésocorticale sous-tendant
les symptômes négatifs (émoussement des affects)
et cognitifs (31). Si l’hypothèse dopaminergique,
bien que non exclusive de toute autre, est celle
qui est étayée par le plus d’arguments, la seule
qui présente des preuves directes (avec les travaux
d’imagerie de Laruelle) et surtout celle qui reste
la mieux vérifiée sur le plan thérapeutique, pour
autant aucune donnée n’indique clairement l’exis-
tence d’anomalies des récepteurs dopaminergiques
chez les patients schizophrènes. En revanche, nous
Glutamate et effets psycho-comportementaux
Le glutamate, en particulier via les récepteurs NMDA, régule
négativement un certain nombre de dimensions psycho-
comportementales comme l’émotion, l’humeur, les capacités
de défense vis-à-vis du stress et de l’anxiété. L’augmentation
anormale des concentrations en glutamate, observée au cours
de la maladie d’Alzheimer, pourrait contribuer à l’apparition
des troubles psycho-comportementaux que l’on y constate, et
la modulation de la transmission glutamatergique pourrait
contribuer à faire régresser certains de ces symptômes, en
particulier les réactions anxieuses et agressives.
R. Bordet

La Lettre du Neurologue • Vol. XIV - n° 11 - décembre 2010 | 411
avons vu que l’activation des récepteurs D2 localisés
sur le versant présynaptique de la synapse gluta-
matergique régule négativement la transmission
glutamatergique cortico-striale mais également
cortico-limbique (9). Cette notion suggère l’impli-
cation du glutamate dans la genèse des symptômes
de la schizophrénie, aussi bien en ce qui concerne les
troubles cognitifs que la production délirante (27).
L’implication du glutamate repose également sur la
capacité de molécules antagonistes des récepteurs
glutamatergiques NMDA, la phencyclidine ou la
kétamine, à induire à doses subanesthésiques des
symptômes positifs (illusions et distorsions percep-
tives, etc.) mais également des signes négatifs (ralen-
tissement, émoussement des affects), des signes
cognitifs (troubles du rappel immédiat et différé,
troubles de la fluence verbale, par exemple) [27]. Il
est frappant de constater que, malgré le caractère
ubiquitaire des récepteurs du glutamate, une
administration unique de cet antagoniste est capable
de reproduire sélectivement des troubles semblables
à ceux décrits dans la schizophrénie (27).
L’existence d’une hérédité dans la schizophrénie est
connue depuis longtemps. Aussi, plusieurs études
génétiques de liaison ont cherché à identifier des
gènes candidats en rapport avec le fonctionnement
des récepteurs du glutamate. Plusieurs gènes codant
pour des protéines interagissant à différents niveaux
avec les récepteurs du glutamate ont ainsi été
impliqués dans la genèse de la schizophrénie. Ces
protéines modifient les propriétés cinétiques, la
disponibilité synaptique, les voies de transduction
intracellulaire du signal ou les phénomènes de
plasticité liés aux récepteurs NMDA (32). Enfin,
l’implication du système glutamatergique dans la
physiopathologie de la schizophrénie s’intègre de
façon intéressante avec la théorie neurodéveloppe-
mentale de la schizophrénie soutenue par plusieurs
équipes.
Plusieurs essais cliniques contrôlés versus placebo
réalisés avec des molécules agissant directement ou
indirectement au niveau du site glycine (indispen-
sable à l’activation du récepteur NMDA, potentiali-
sation de l’action du glutamate) ont d’ailleurs montré
une diminution de la symptomatologie négative ou
une amélioration des fonctions cognitives chez des
patients schizophrènes poursuivant leur traitement
antipsychotique habituel (33). En conclusion, la
mise en évidence de l’implication du glutamate
dans la schizophrénie a complètement bouleversé
les concepts psychopathologiques relatifs à cette
maladie mais également l’approche neurochimique
des symptômes (2).
Conclusion
Nous avons vu que l’essentiel des fonctions
cérébrales est sous la dépendance du glutamate
et que de nombreuses maladies neurologiques
résultent d’un dysfonctionnement de cette trans-
mission. Toutefois, plusieurs questions restent
encore en suspens, notamment celle de la place
du glutamate dans le vieillissement cérébral normal.
En effet, les fonctions cognitives et la motricité
s’altèrent avec l’âge, sans qu’il y ait de processus
pathologique sous-jacent. Les études portant sur le
lien entre le glutamate et le vieillissement cérébral
sont complexes en raison de l’hétérogénéité des
modèles et de l’absence de marqueur fiable du
processus de vieillissement (34). La seconde
question en suspens concerne les débouchés
pharmacologiques et thérapeutiques des données
recueillies sur l’implication du glutamate dans ces
différents processus pathologiques. Comme nous
l’avons vu, des résultats prometteurs ont déjà été
obtenus dans de nombreuses indications. Toutefois,
en raison du caractère ubiquitaire de la répartition
des synapses glutamatergiques dans le système
nerveux central, il est très probable que l’efficacité
d’une démarche thérapeutique soit conditionnée par
l’intégration de l’ensemble des facteurs d’interaction
existant entre le glutamate, les différents systèmes
neuronaux et les autres neurotransmetteurs. ■
 6
6
1
/
6
100%