Composés halogénés et organométalliques

CH 7. Composés halogénés
et organométalliques.
SUBSTITUTION ET ELIMINATION

RX
Partie organique Halogène
Les haloalcanes constituent la plaque tournante de la
synthèse organique.

CH 7. Composés halogénés et organométalliques.
SUBSTITUTION ET ELIMINATION
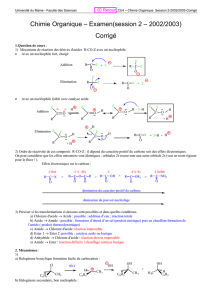
1. SN : la substitution nucléophile

La réaction est en principe réversible :
le groupe nucléofuge est aussi un nucléophile.
CH3CH2Br Br-
OH
-
CH3CH2OH
X-
RNu
RX
-Nu :
Br-
CH3CH2Br CH3CH2NH3
NH3
+
Nu : RXRNu+X-
Un groupe sortant, nucléofuge, est remplacé par
un groupe entrant, nucléophile, chargé ou non.

2. Substitutions nucléophiles sur halogénoalcanes
1. SN : la substitution nucléophile
CH 7. Composés halogénés et organométalliques.
SUBSTITUTION ET ELIMINATION
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
1
/
65
100%