1.2 Insuffisance cardiaque due à une pathologie cardiaque

Feat.Sovietik
PAT – IC 1/10
Insuffisance cardiaque
I. Rappel physiopathologique
1. Définition et origines
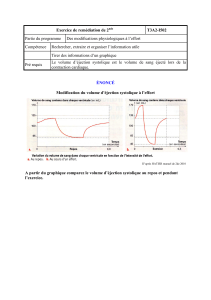
Insuffisance cardiaque : incapacité du myocarde à éjecter suffisamment de sang lors de chaque
systole => les besoins en oxygène de l’organisme ne sont pas satisfaits.
Pour une même pression de remplissage diastolique, le débit systolique se réduit de plus en plus en
fonction du degré de gravité de la pathologie.
On quantifie l’insuffisance cardiaque par :
La fraction d’éjection : % de volume ventriculaire éjecté lors de la systole.
A l’état normal : fraction d’éjection : au moins 60% pour un cœur sain
Forme grave d’insuffisance cardiaque : 20 à 30%
L’insuffisance cardiaque affecte principalement le ventricule gauche.
Etiologies : HTA, maladies coronaires (athérosclérose), cardiomyopathies, insuffisance aortique,
réduction du myocarde fonctionnel par exemple après un infarctus.
2 types d’insuffisance cardiaque :
- associée à une diminution de volume d’éjection systolique = insuffisance systolique.
Observée suite à une surcharge chronique de volume ou de pression ou suite à une maladie
cardiaque.
- associée à un empêchement de remplissage diastolique = insuffisance diastolique.
Observée suite à une augmentation chronique de rigidité de la paroi du ventricule.
Insuffisance systolique : Volume d’éjection systolique et débit cardiaque ne sont pas en adéquation
avec les besoins de l’organisme.
Insuffisance diastolique : Débit cardiaque ne peut être ajusté que par une augmentation de la
pression de remplissage diastolique.
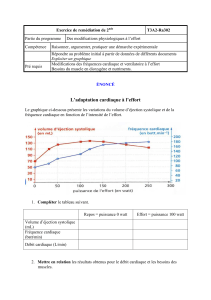
Initialement l’insuffisance cardiaque ne se révèle que lors du travail intense. Puis l’insuffisance
cardiaque se développe au cours du temps : observation lors d’effort de tous les jours puis même
au repos.
1.1 Insuffisance cardiaque due à une surcharge en volume
En cas d’insuffisance aortique ou mitrale : un volume supplémentaire de sang va faire des
allers-retours entre l’oreillette et le ventricule. Il va s’additionner au volume d’éjection systolique
(= surcharge de volume). Le volume diastolique et le rayon du ventricule gauche vont augmenter =>
augmentation de la tension pariétale. La force développée par le cœur pour l’éjection du volume
systolique normal devra être augmentée => la consommation en oxygène du cœur augmente.
A long terme : le cœur ne peut pas satisfaire ses besoins accrus en oxygène, il en résulte :
diminution du volume d’éjection systolique => diminution du débit cardiaque => diminution de la
pression artérielle

PAT – Insuffisance cardiaque 2/10
1.2 Insuffisance cardiaque due à une pathologie cardiaque
En cas d’infarctus du myocarde ou d’affection des coronaires la fraction du myocarde
fonctionnel est diminuée => la contractilité du myocarde diminue. Il en résulte une augmentation
des volumes systoliques et diastoliques et une diminution du volume d’éjection systolique.
En cas de cardiomyopathie :
- dilatative : on observe une dilatation du ventricule conduisant à une surcharge de volume
- hypertrophiée : on observe un défaut de remplissage du ventricule car le muscle est moins
élastique, ce qui rend l’éjection difficile.
1.3 Insuffisance cardiaque due à une surcharge de pression
Dans le cas d’HTA ou de sténose de l’aorte, la tension pariétale du ventricule gauche va être
augmentée car la pression intra ventriculaire nécessaire pour éjecter le sang sera plus grande
développement d’une insuffisance systolique avec diminution de la contractilité, ce qui
entraîne un risque d’hypertension pulmonaire
2. Conséquences structurales du cœur
Le cœur est donc soumis à une surcharge chronique de volume ou de pression => le cœur
s’hypertrophie.
Il existe plusieurs facteurs impliqués dans le remodelage du myocarde :
- augmentation de la tension pariétale => augmentation de la concentration cytosolique de
calcium
- facteurs mitogéniques d’origine systémique (catécholamines, arginine vasopressine,
angiotensine II) qui vont favoriser la prolifération cellulaire et la synthèse de matrice
extracellulaire par les cellules du myocarde.
- facteurs mitogéniques produits localement dans muscle cardiaque (endothélium, facteur
activateur des plaquettes : PDGF, facteur de croissance des fibroblastes : FGF) qui vont
stimuler la prolifération de cellules du myocarde et la synthèse de matrice extra cellulaire
impliquées dans l’hypertrophie.
De plus, on observe chez l’insuffisant cardiaque une diminution de la formation de facteurs
vasoprotecteurs dérivés de l’endothélium : prostacycline (PGI2), NO.
Au final, la cellule myocardique s’agrandit et ses propriétés changent : elle devient réfractaire aux
catécholamines (diminution du nombre de récepteur β1) et on observe un découplage entre
récepteur et protéine G.
L’augmentation de masse muscle cardiaque favorise initialement l’adaptation du cœur à
une surcharge de pression ou de volume. Si paroi du ventricule s’épaissit, on observe une
diminution de tension pariétale et donc une diminution de la consommation en oxygène du cœur.
Dans cette phase initiale : le débit cardiaque n’est pas obligatoirement diminué : le muscle
cardiaque essaye de lutter.
Les symptômes cliniques de l’insuffisance cardiaque sont surtout la conséquence d’un
dysfonctionnement diastolique car l’hypertrophie musculaire entraîne une rigidité du ventricule. Le
ventricule se dilate moins.
Le remplissage du ventricule gauche ne pourra se faire normalement qu’à de fortes
pressions. Ces fortes pressions ont un effet délétère en augmentant la consommation en oxygène
du cœur nécessaire pour dilater le ventricule.

PAT – Insuffisance cardiaque 3/10
Les symptômes sont alors :
- dyspnée
- augmentation de la pression veineuse
- stase veineuse
- oedème pulmonaire et périphérique
- cardiomégalie (= gros cœur fortement hypertrophié)
Symptômes mineurs : arythmie, tachycardie, toux nocturne, augmentation de la taille du foie
3. Conséquences biologiques et hormonales
L’organisme essaye par des mécanismes réflexes neuro-humoraux de contrecarrer l’évolution
de la maladie, mais il va s’établir un cercle vicieux qui contribue au développement de la
pathologie :
Activation du système sympathique : il en résulte une réduction de la pression partielle en
oxygène et une réduction de la pression artérielle car le débit cardiaque diminue. Les
chémorécepteurs et les barorécepteurs vont être stimulés, ce qui entraîne successivement :
activation réflexe du système sympathique
libération de catécholamines
augmentation de la fréquence et de la force de contraction qui permettent d’éviter au moins
au repos la chute brutale et dangereuse du débit cardiaque.
Augmentation du tonus vasculaire par l’activation des récepteurs α1, ce qui potentialise le
retour veineux et contribue à l’augmentation de la pression de remplissage ventriculaire et à induire
la dilatation du cœur. Cette dilatation initiale du ventricule permet d’augmenter le volume
d’éjection systolique à l’aide du mécanisme de Frank Starling (l’étirement du muscle jusqu’à un
point optimal permet une meilleure contraction ; pour n’importe quel tissu il faut un étirement
optimal pour avoir une contraction optimale).
Ce mécanisme d’adaptation très vite dépassé. Une fois dépassé, on observe une forte augmentation
de la tension pariétale et de la fréquence cardiaque conduisant à une consommation excessive
d’oxygène.
De plus, l’activation réflexe du sympathique entraîne une augmentation de la résistance vasculaire
périphérique (action des récepteurs α1 des vaisseaux), d’où :
augmentation de la post charge
diminution du volume d’éjection systolique
Le cœur affaibli perd la capacité de maintenir le volume d’éjection constant quand la post charge
est modifiée.
Activation du système rénine angiotensine aldostérone : il en résulte une diminution du débit
cardiaque et activation du système sympathique permettant la redistribution de la perfusion des
organes dans le but de maintenir une perfusion du cerveau et des coronaires au dépend du muscle
squelettique. On observera des signes de fatigue musculaire, une diminution de la perfusion de la
peau (d’où un teint pâle), et du rein : réduction de la pression de filtration glomérulaire
stimule la sécrétion de rénine par appareil juxta glomérulaire.
formation accrue d’angiotensine II = acteur majeur de l’insuffisance cardiaque.
L’angiotensine II :
- induit une vasoconstriction => augmentation de la pré et de la et de la post charge.
- est un puissant facteur mitogénique augmentation de l’hypertrophie cardiaque

PAT – Insuffisance cardiaque 4/10
- active la sécrétion d’aldostérone et d’ADH rétention hydrosodée hausse du volume
extracellulaire (= volume du plasma) retour veineux plus intense on favorise au départ
l’adaptation de Frank Starling.
La sécrétion d’ADH est également stimulée par l’hypotension.
L’étirement des oreillettes stimule la sécrétion de peptide natriurétique auriculaire qui augmente
l’élimination de sodium et d’eau et compense partiellement l’action de l’Angiotensine II.
A long terme, l’activation du SRAA va contribuer au développement d’œdèmes.
CCL : Ces mécanismes régulateurs vont provoquer une augmentation progressive de la pression de
remplissage ventriculaire qui, par l’intermédiaire de l’oreillette, se transmet au système veineux. Si
l’insuffisance cardiaque est au niveau du cœur gauche il y aura des stases pulmonaires avec
exsudation vers le milieu extracellulaire. Si elle est au niveau du cœur droit il y aura des stases dans
la grande circulation.
L'excès de pression fait sortir le fluide des capillaires vers le tissu interstitiel et vers les alvéoles.
Un œdème pulmonaire induit une dyspnée et une tachypnée avec réduction des échanges gazeux
quand la pathologie est avancée, avec hypoxie voire hypercapnie systémique.
II. Les principes généraux du traitement
Le but de la thérapie est de rétablir le débit cardiaque en augmentant le volume d’éjection
systolique et d’essayer d’éliminer les symptômes de congestion pulmonaire.
3 approches pharmacologiques :
1. Diminution de la pré-charge et de la post-charge :
- diminution de la pré-charge : on va dilater les vaisseaux, ce qui entraîne une réduction du
retour veineux au cœur. Conséquence : moindre remplissage du ventricule gauche, donc
diminution de la pression ventriculaire et diminution des symptômes de congestion dans le
système pulmonaire.
- diminution de la post-charge : on utilise surtout des agents qui dilatent les vaisseaux de
résistance. Le but est de faciliter l’éjection du sang durant la systole. Cela entraîne une
amélioration de la fraction d’éjection avec réduction du volume diastolique du ventricule
gauche au repos. Il y a normalisation de la pression diastolique qui était pathologiquement
élevée.
Les principaux médicaments utilisés pour réduire :
- la pré- et post-charge →IEC, sartans, -bloquants
- préférentiellement la pré-charge → dérivés nitrés.
2. Utilisation de diurétiques :
Pour réduire le volume sanguin circulant afin d’améliorer les symptômes de la congestion.
Conséquence : on aura un remplissage ventriculaire plus faible. On va soulager le cœur en réduisant
la pré-charge.
Les diurétiques de l’anse ont la capacité de réduire le tonus des vaisseaux de capacitance (veines) =
effet thérapeutique recherché dans l’œdème pulmonaire aigu.
Avant l’installation de la diurèse, on assiste à un déplacement des fluides du poumon dans la
circulation veineuse avec réduction de la pression de remplissage du ventricule gauche.

PAT – Insuffisance cardiaque 5/10
3. Utilisation de substances inotropes + (digitaliques) :
Cela améliore la fraction d’éjection et la pression diastolique qui était pathologiquement élevée
dans le ventricule gauche.
CCL : le but final du traitement de l’IC est d’améliorer les symptômes et l’hémodynamique à court
terme, et de réduire la mortalité (donc effet préventif). Les molécules utilisées actuellement n’ont
pas montré une diminution de la mortalité sauf les IEC, sartans et -bloquants.
III. IEC / Sartans
1. IEC :
Actuellement tout insuffisant cardiaque doit recevoir un IEC. C’est le traitement de base de l’IC,
utilisé seul ou en association. C’est le traitement de base, car le système Rénine-Angiotensine a un
rôle déterminant dans l’évolution de la pathologie.
Propriétés bénéfiques :
- réduction de la formation d’Angiotensine II : donc diminution de la vasoconstriction, diminution
des effets mitogènes et de la synthèse de la matrice extracellulaire. Et secondairement : moins
d’aldostérone et d’arginine vasopressine, donc moins de rétention hydrosodée, et diminution
du tonus sympathique.
- empêche la dégradation des kinines, donc il y aura plus de bradykinine qui va dilater les
vaisseaux.
Sur le plan hémodynamique :
- diminution de la résistance vasculaire périphérique. Donc diminution de la post-charge et donc
meilleure vidange ventriculaire.
- diminution de la pression capillaire pulmonaire
- augmentation du débit sanguin rénal et de la filtration glomérulaire.
- effet bénéfique sur le remodelage ventriculaire.
Les IEC sont capables de diminuer la mortalité : effet bénéfique d’environ 25%. Cet effet
bénéfique se maintient souvent au cours du traitement sur plusieurs années. De plus, ils améliorent
les symptômes, la tolérance à l’effort et la qualité de vie du patient.
Souvent on utilise les posologies maximales tolérées. La surveillance clinique regarde
l’évolution des symptômes de l’IC, les variations de la pression artérielle, s’il y a hypotension
orthostatique, … (attention surtout chez les sujets âgés, en cas de co-prescription d’un diurétique
ou d’un autre vasodilatateur)
La surveillance biologique consiste en : bilan sanguin, kaliémie et créatininémie : à vérifier de
manière régulière tout au long du traitement (on conseille une fois par mois au départ puis une fois
par an).
Certains patients ont une toux sèche rebelle avec les IEC, on utilise alors les sartans.
2. Sartans :
Ils sont capables de diminuer la mortalité associée à l’IC.
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%