Contrôle continu : Evolution Moléculaire (EM7BMAAM

1
Contrôle continu : Evolution Moléculaire (EM7BMAAM) – Octobre 2015
Questions de cours :
1. Quel est l'impact sur le calcul de la distance évolutive entre deux séquences, de la présence de
substitutions multiples ayant pu se produire au même site ? Comment palie-on à ce problème ?
( 2 points)
Les substitutions multiples qui ont pu se produire au même site ne sont pas directement
observables par la comparaison des séquences. Leur présence résulte en une sous-estimation des
distances évolutives entre les séquences analysées. Ce phénomène est plus critique dans le cas des
séquences d’acides nucléiques car elles possèdent un alphabet plus pauvre que les séquences
protéiques : quatre lettres au lieu de 20.
Pour tenter de corriger le biais du aux mutations multiples, des hypothèses sont faites sur la
façon dont les bases ou les acides aminés se sont substitués à un locus donné au cours de
l'évolution conduisant à la construction d'un modèle évolutif. Plusieurs modèles évolutifs
existent pour analyser les séquences d'acides nucléiques comme pour analyser les séquences
protéiques. Ces modèles vont de modèles simples, ayant une vision simplificatrice de l'évolution
à des modèles de plus en plus complexes rendant mieux compte de l'évolution. Cependant, les
modèles plus complexes nécessitent l'estimation d'un nombre plus grand de paramètres et ne
peuvent être utilisés que si le jeu de données est suffisamment grand (nombre de séquences et
taille des séquences), sinon l'estimation des paramètres risque d'être erronée, ce qui est pire que
l'utilisation d'un modèle évolutif plus simple quant à la reconstruction phylogénétique obtenue.
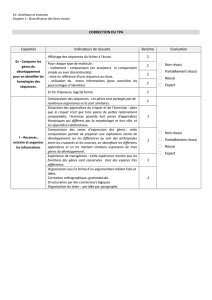
2. Le tableau suivant montre la distribution de 4 caractères dans 4 organismes différents.
Quelle méthode de reconstruction d'arbre utiliseriez-vous pour établir les relations
phylogénétiques existant entre ces espèces? (1 point)
Nous avons ici des données morphologiques représentées par la présence/absence d'un
caractère dérivé. L'approche que nous utiliserons sera donc une approche cladistique
utilisant la méthode de parcimonie pour la construction de l'arbre phylogénétique.
Reconstruisez cet arbre en expliquant les principes de la construction et en détaillant chaque
étape.
Caractères
Gésier (1)
Membrane
nictitante (2)
Plume (3)
Sang chaud
(4)
Espèces
Grenouille
0
0
0
0
Chien
0
0
0
1
Alligator
1
1
0
0
Pie
1
1
1
1
0 signifie que le caractère n’est pas observé et 1 que le caractère est observé.
Reconstruction de l'arbre : (2 points)
Première étape : On construit un arbre avec les 3 premières espèces et on reporte sur les branches le
numéro du caractère transformé. Ici la grenouille sert de groupe externe car elle ne possède aucun des
caractères dérivés analysés. Nous obtenons l'arbre présenté ci-dessous.

2
Seconde étape : Nous allons rajouter la quatrième espèce sur cet arbre. Il y a trois possibilités car trois
branches internes. Pour chaque arbre, nous allons placer sur ces branches l'apparition des caractères
dérivés permettant d'expliquer la topologie. Nous conserverons l'arbre le plus parcimonieux, c'est-à-
dire celui dont la topologie s'explique par le minimum de changements. Dans note cas, il s'agit de
l'arbre encadré dont la topologie s'explique par 5 changements. Pour les deux autres arbres, nous avons
supposé que les caractères 1 et 3 étaient apparus indépendamment sur les branches menant à l'alligator
et à la pie. Nous aurions pu faire l'hypothèse qu'ils étaient apparus avant la séparation
chien/alligator(pie) et auraient subi une réversion sur la branche menant à au chien. Le nombre de
changement aurait été équivalent.
Problème (basé sur les travaux publiés de Aoki et al., Mol. Biol. Evol. 30(11): 2494-2508, 2013)
Plusieurs espèces bactériennes appartenant au groupe taxonomique des -protéobactéries et certaines
appartenant au groupe des -protéobactéries ont établi une relation symbiotique avec les
légumineuses. Ces espèces ont été appelées-rhizobia et -rhizobia. Cependant l'origine évolutive des
gènes de nodulation impliqués dans la fixation de l'azote reste incertaine. Dans cet article, les auteurs
se sont intéressés à l'origine et l'évolution de deux de ces gènes, nodI et nodJ, dont les produits
protéiques jouent un rôle clef dans la sécrétion des facteurs Nod qui sont reconnus par les
légumineuses lors de la nodulation.
Ils ont recherché les protéines présentant des similarités de séquences avec les protéines NodI et NodJ
dans les génomes de bactéries et d'archés complètement séquencés. Les résultats de cette recherche
ont montré que les protéines NodI et NodJ présentaient des similarités de séquences avec deux
domaines fonctionnels portés par une même protéine DRA qui est un transporteur ABC appartenant à
la famille des exporteurs impliqués dans la résistance aux drogues et aux antibiotiques. NodI possède
des conservations de séquence avec le domaine ATPase de DRA (DRA-ATPase) et NodJ avec le
domaine perméase (DRA-permease).
A partir de ces résultats, ils ont extrait un jeu de données plus petit en choisissant des espèces
bactériennes représentatives des différents groupes taxonomiques pour réaliser leurs analyses
évolutives.

3
1) Une fois le jeu de données (i.e., les séquences) à analyser établi, décrivez les différentes étapes qui
doivent être réalisées pour construire un arbre phylogénétique. (2 points)
Un alignement multiple sera tout d'abord construit en utilisant un logiciel à cet effet (Muscle,
ClustalO,..). Cet alignement pourra être amélioré par correction manuelle ci-nécessaire. Ensuite,
une recherche du modèle évolutif le mieux adapté aux données sera réalisé à l'aide de Protest
(données de séquences protéiques) ou de JModelTest (données de séquences nucléiques). Le
résultat de ces deux méthodes nous informera aussi sur la nécessité ou non d'utiliser la
correction Gamma permettant de prendre en compte plusieurs classes de vitesse d'évolution des
sites. Pour les modèles évolutifs, dans le cas des séquences protéiques nous disposons notamment
des modèles PAM, JTT, WAG et LG. Dans le cas des séquences nucléiques, un plus grand
nombre de modèles sont disponibles (Jukes et Cantor, Kimura 2 paramètres, Tamura, Tamura
et Nei etc.). Ces modèles tentent de modéliser ce qui est connu sur l'évolution des séquences et
tentent de corriger le biais du aux substitutions multiples que l'on ne peut pas observer
directement à partir des séquences actuelles et qui conduisent à une sous estimation des
distances évolutives.
L'arbre phylogénétique sera construit en utilisant ce modèle et une méthode de maximum de
vraisemblance (PhyML). La robustesse de chacune des branches de la topologie sera évaluée par
l'utilisation de la méthode du bootstrap. Deux arbres pourront être construits pour le même jeu
de données et ceci avec deux méthodes différentes (PhML et BioNJ par exemple). Si les deux
arbres sont congruents, cela renforcera notre confiance dans la topologie obtenue.
Comme les gènes nodI et nodJ sont toujours trouvés en opérons et que les arbres obtenus
indépendamment sur chacune des protéines (NodI et NodJ) présentent quasiment la même topologie,
dans la suite de l'analyse, ils ont concaténé dans une même séquence, les protéines NodI et NodJ
codées par le même opéron. Un premier arbre a été construit en utilisant PhyML (Figure 1).
2) Pourquoi avoir établi l'arbre à partir des séquences protéiques et non à partir des séquences de leurs
gènes ? (0,5 point)
Les arbres ont été construits à partir de séquences protéiques et non nucléiques car lorsque les
espèces sont distantes dans l'évolution, les séquences nucléiques peuvent avoir subi des
substitutions multiples qui conduiront à une sous-estimation de leurs distances évolutives. On
peut même dans certains cas avoir perdu le signal phylogénétique. On préfère donc travailler au
niveau protéique.
3) A quelle classe de méthodes de reconstruction d'arbre appartient PhyML ? (0,5 point)
PhyML méthode du maximum de vraisemblance
4) A quoi correspondent les nombre figurant sur les branches des arbres des Figures 1 et 2 (0,5
point)? Comment sont-ils obtenus (1 point) ? Quel est l'intérêt de calculer ces valeurs ? (0,5 point)
Les nombres sur les branches correspondent aux valeurs de bootstrap.
Ils sont obtenus par ré-échantillonnage par tirage aléatoire avec remise des positions alignées
pour construire un alignement de même longueur. Un arbre phylogénétique sera construit en
utilisant cet alignement aléatoire. Ce processus est réitéré plusieurs fois (100 au minimum).
Ensuite, le nombre de fois où chaque branche de la topologie de départ est retrouvée dans ces
arbres "aléatoires" est calculé. La valeur de bootstrap correspondra au pourcentage de fois où
la branche a été retrouvée.
Cette méthode permet de tester individuellement la validité de chaque branche interne de
l’arbre et d'estimer sa robustesse. De manière générale, une faible valeur de bootstrap indique
que la quantité d’information supportant la bipartition induite par une branche interne est
faible. Si on applique les critères standards utilisés en statistique, il ne faudrait considérer
comme robuste que les branches ayant un support de bootstrap ≥ 95%. Des travaux ont montré
que ce seuil était trop élevé et que des supports de 70% pouvaient correspondre à des branches
valides.

4
5) Pourquoi avoir utilisé pour la construction de cet arbre la séquence de Methylococcus capsulatus
Bath qui une bactérie n'appartenant ni au - ni au -protéobactéries ? (0,5 point)
Cette séquence joue le rôle de groupe externe. Elle permet donc d'enraciner notre arbre, c'est-à-
dire d'identifier le nœud racine correspondant à l'ancêtre hypothétique de nos séquences
d'intérêt, à savoir des - et - protéobacteries
6) Parmi les espèces présentes sur l'arbre, qu'elles sont celles dont le génome renferme à la fois les
gènes codant pour NodI, NodJ et DRA-ATPase/permease ? (1 point)
Les espèces dont les génomes renferment à la fois les gènes pour NodI, NodJ et DRA-
ATPase/permease sont :
Burkholderia sp CCGE1002
Burkholderia phymatum STM815
7) Parmi les espèces présentes sur l'arbre, qu'elles sont celles dont le génome renferme uniquement les
gènes codant pour NodI, NodJ ? (0,5 point)
Toutes les espèces (13 au total) appartenant au groupe des -rhizobia.
8) Etablir un scénario évolutif pour expliquer l'origine des gènes nodI et nodJ dans les - et -rhizobia
(Pour répondre à cette question, vous vous attacherez à décrire les évènements de duplication, perte de
gènes et transferts horizontaux qui ont pu se produire.) (2 points)
Les différents arbres sont enracinés par des groupes externes. Il est donc possible d'orienter les
événements évolutifs à partir des branches les plus profondes de l'arbre. Chez les
Burkholderiaceae, seuls les génomes de Burkholderia phymatum STM815 et de Burkholderia sp
CCGE1002 possèdent les gènes codant pour NodI/NodJ en plus du gène codant pour DRA, ce
qui suggère qu'une duplication de gène s'est produite chez leur dernier ancêtre commun, ancêtre
des Burkholderia. Sous cette hypothèse l'absence des gènes nodI et nodJ, chez les 5 autres
Burkholderia, peut s'interpréter comme une délétion dans ces 5 génomes (figure 1). Les régions
codantes fusionnées dans DRA se sont séparés en deux gènes (fission) après la duplication.
L'arbre enraciné de la figure 2 montre que les -rhizobia et les-rhizobia forment deux clades
bien distincts supportés par une valeur de bootstrap de 100. Le gène codant pour DRA est
absent des génomes d'-rhizobia. En l'absence d'information sur la présence de ce gène chez les
-protéobactéries, groupe auquel appartiennent les -rhizobia, il n'est pas possible de conclure
sur l'absence de ce gène dans ces génomes. La localisation des gènes nodIJ des -rhizobia
comme sous-arbre frère du sous-arbre nodIJ des Burkholderia dans la figure 1 suggère
fortement (valeur de bootstraps 100%) que ces gènes ont été acquis par transfert horizontal
dans le génome ancêtre à l'ensemble des -rhizobia actuelles et ceci à partir d'un ancêtre des
Burkholderia actuelles.
Ils ont poursuivi leur étude en construisant un arbre phylogénétique des espèces bactériennes en
utilisant les séquences de 25 familles de protéines présentes dans l'ensemble des génomes étudiés. Ils
ont ensuite comparé la topologie des deux arbres, celui des espèces bactériennes et celui des protéines
NODIJ/DRA-ATPase/permease (Figure 2).
9) Pour construire l'arbre des espèces bactériennes, les gènes codant pour les protéines constituant
chacun des 25 groupes de séquences doivent-ils être homologues, paralogues ou orthologues ? (1
point)
Les séquences appartenant à chacun des groupes doivent être orthologues et donc également
homologues.
10) Quand l'arbre phylogénétique des espèces et l'arbre des protéines étudiés sont congruents, quelle
hypothèse peut-on faire quant à la façon dont les gènes codant pour les protéines ont été hérités par les
différents génomes ? (0,5 point)

5
Dans ce cas, les gènes ont été hérités verticalement. C'est-à-dire que le gène était présent dans le
génome de l'espèce ancêtre et a été hérité par les espèces filles suite au processus de spéciation.
11) Les deux arbres obtenus ((espèces et protéines NODIJ/DRA-ATPase/permease) sont-ils
congruents ? Argumentez votre réponse. (2 points)
Non ces deux arbres ne sont pas congruents car la comparaison avec l'arbre des espèces montre
des lignes qui se croisent indiquant que des feuilles dans les deux arbres ne sont pas localisées au
même endroit dans chacune des deux topologies.
En fait, si nous distinguons les deux sous-arbres, le sous-arbre correspondant à l'évolution de la
protéine DRA-ATPase/permease est congruent avec le sous-arbre espèces des -protéobactéries,
révélant que ce gène a été transmis de façon verticale au sein de ces espèces (à travers le
processus de spéciation).Par contre, le sous-arbre correspondant à l'évolution de la protéine
NodIJ n'apparaît pas congruent avec le sous-arbre espèces des -rhizobia. En effet, les
séquences NodIJ des espèces Ensifer fredii NGR234, Rhizobium etli CFN 42, Rhizobium etli CFN
652, Mesorhizobium loti MAFF303099, Mesorhizobium ciceri biovar biserrulae WSM1271, Ensifer
meliloti 1021 et Ensifer medicae WSM419 ne sont pas localisées au même endroit dans les deux
topologies (arbre séquences NodIJ et arbre espèces) indiquant que les gènes nodI et nodJ de ces
espèces ont été acquis par transferts horizontaux. Cependant, ces observations sont à pondérer
en raison des valeurs très faibles de bootstrap observées sur les branches de l'arbre NodIJ.
12) Les auteurs ont montré que les longueurs des branches du sous-arbre des protéines NodI/J étaient
statistiquement supérieures à celles du sous-arbre DRA-ATPase/permease. Quelle interprétation
biologique pouvez-vous apporter à ce résultat ? (1 point)
Les longueurs des branches d'un arbre étant proportionnelle au nombre de mutations observé
entre les séquences, un accroissement de cette longueur indique une augmentation du nombre de
mutations entre les séquences comparées, donc une accélération de la vitesse d'évolution. Ceci
est fréquemment rencontré après un évènement de duplication d'un gène, où la nouvelle copie
du gène n'est pas soumise à la contrainte évolutive et peut donc accumuler des mutations sans
que cela ait un impact sur la fitness de l'organisme (le gène "n'a pas encore de fonction").
L'accumulation de ces mutations permet ainsi l'apparition d'une nouvelle fonction qui pourra
être sélectionnée par l'évolution.
13) Le scénario évolutif largement décrit dans la littérature considère que les -protéobactéries sont à
l'origine des rhizobias. Pensez-vous que les analyses évolutives réalisées sur les gènes nodI et nodJ
conforte cette hypothèse ? Justifier votre réponse. (1,5 point)
Non, les résultats obtenus par cette analyse évolutive ne conforte pas ce scénario évolutif. En
effet, il est montré ici que les gènes nodI et nodJ seraient apparus dans les Burkholderia, donc
dans les -protéobacteries, à la suite d'une duplication du gène ancêtre codant la protéine DRA-
ATPase/permease dans le génome ancêtre des espèces actuelles de Burkholderia et que les -
rhizobia auraient acquis ces gènes par transfert horizontal (HGT). Nous pouvons noter que les
conclusions reposent sur un faible jeu de données et que des analyses ultérieures reposant sur
une plus grande diversité de génomes seraient requises pour confirmer ou modifier le modèle
évolutif proposé.
1
/
5
100%