REVUE DE PRESSE

REVUE DE PRESSE dirigé par
le Pr P. Bouche
122 | La Lettre du Neurologue Nerf & Muscle • Vol. XIV - n° 4 - avril 2010
Peut-on différencier une polyradiculonévrite aiguë
de type Guillain-Barré d’une forme à début aigu
de polyradiculonévrite chronique ?
Les auteurs ont réalisé une étude rétrospective (de 1993 à 2007) à partir des dossiers de patients
ayant une polyradiculonévrite aiguë de type Guillain-Barré (AIDP) et des formes de polyradicu-
lonévrites chroniques à début aigu (A-CIDP). L’étude a été menée au Health Sciences Centre de
London dans l’Ontario (Canada) par l’équipe d’Angelika Hahn. Les diagnostics ont été réalisés
selon les critères actuellement acceptés: pour l’AIDP, l’installation des symptômes doit se faire
en moins de 4 semaines et, pour l’A-CIDP, elle est également de moins de 4 semaines mais la
neuropathie continue de progresser au-delà de 8 semaines, ou il existe plus d’une rechute après
amélioration ou disparition des symptômes, ou le maintien du traitement est nécessaire avec
plus d’une série supplémentaire d’Igi.v., d’échanges plasmatiques ou d’immuno suppresseurs.
Les paramètres cliniques retenus pour comparer les 2 groupes étaient une ataxie sensitive,
une altération marquée de la sensibilité vibratoire, un déficit sensitif superficiel en gants et
chaussettes, des symptômes sensitifs marqués, des signes dysautonomiques, une ventila-
tion mécanique, une faiblesse des muscles respiratoires, une paralysie faciale, une atteinte
bulbaire, des douleurs dorsales ou radiculaires et des antécédents d’une maladie infectieuse.
Les paramètres électrophysiologiques étaientau nombre de 3: l’épargne du potentiel sensitif
du nerf sural, le rapport sensitif supérieur à 1 et la présence d’ondes A au cours de l’étude des
ondes F. L’épargne du potentiel sensitif du sural par rapport à l’altération des nerfs médian
et/ou radial est considérée comme très évocatrice d’AIDP. Le rapport sensitif consiste à calculer
le rapport des amplitudes des nerfs sural + radial sur cubital + médian. Les résultats ont mis
en évidence: 15 patients avec A-CIDP et 30 avec AIDP. Sur le plan clinique, les paramètres
qui diffèrent de façon significative sont, en faveur d’A-CIDP, l’ataxie sensitive et, pour l’AIDP,
l’atteinte dysautonomique et la précession d’une affection infectieuse. En ce qui concerne les
paramètres électrophysiologiques, aucun des 3 paramètres retenus ne montre de différence
significative entre les 2 groupes. Les auteurs concluent en soulignant que la distinction entre
A-CIDP et AIDP est initialement faite uniquement sur des éléments cliniques.
P. Bouche,
hôpital de la Pitié-Salpêtrière, Paris
Caractéristiques cliniques et pronostic
des myasthénies oculaires du sujet âgé
Dans la population générale, l’incidence annuelle de la myasthénie est de 21,2 pour 1 million.
La récente augmentation de l’incidence montre cependant que celle-ci affecte essentiel-
lement les sujets dont les symptômes commencent après l’âge de 70 ans. Toutes classes d’âge
confondues, la myasthénie, dont les premiers symptômes sont purement oculaires, touche 39
à 53 % des patients, évoluant vers une myasthénie généralisée dans 50 % à 80 % des cas.
J.A. Allen et al. rapportent les résultats d’une étude réalisée à Boston entre 1980 et 2007,
afin de préciser les relations entre la myasthénie oculaire à début tardif, son diagnostic, son
évolution et son pronostic avec la mise sous traitement immunomodulateur. Les critères d’in-
clusion étaient, en plus de la symptomatologie clinique évocatrice, au moins un des critères
suivants: un taux positif d’anticorps AChR, un décrément de plus de 10 % au cours de la
stimulation répétitive, un jitter positif à l’étude en fibre unique, une réponse clinique certaine
à la pyridostigmine et/ou à l’édrophonium. La sévérité de la myasthénie était évaluée selon
les critères de la Myasthenia Gravis Foundation of America (MGFA): classe I, patients dont la
maladie est limitée à la sphère oculaire. Les résultats sont les suivants: sur les 91 patients de
la base de données de seniors, les formes oculaires pures représentaient 39 cas (43 %), dont
Commentaire
Il s’agit d’un article bien décevant venant de la
part d’une telle équipe. Il y a pourtant un intérêt
certain à différencier les formes à début aigu des
A-CIDP des vrais syndromes de Guillain-Barré, l’at-
titude thérapeutique est alors différente car le
traitement par Igi.v. (ou autre thérapeutique à
visée immunitaire) doit être prolongé. Si les critères
cliniques de différenciation paraissent corrects et
ne montrent en fait que ce que l’on attendait,
c’est-à-dire la plus grande fréquence d’atteinte
dysautonomique et respiratoire dans le syndrome
de Guillain-Barré et de maladies infectieuses préa-
lables, et, dans les A-CIDP, des formes plus souvent
sensitives ataxiantes, ce sont les paramètres
électro physiologiques qui interpellent. On aurait
aimé voir pris en compte d’autres paramètres
tels que la dispersion des réponses motrices, la
présence de blocs de conduction, les amplitudes
motrices distales et les ondes F. Une étude plus
consistante est ainsi nécessaire et souhaitable, car
l’enjeu paraît d’importance.
Référence bibliographique
Dionne A, Nicolle MW, Hahn AF. Clinical and electrophy-
siological parameters distinguishing acute-onset chronic
inflammatory demyelinating polyneuropathy from acute
inflammatory demyelinating polyneuropathy. Muscle
Nerve 2010;41:202-7.

REVUE DE PRESSE dirigé par
le Pr P. Bouche
124 | La Lettre du Neurologue Nerf & Muscle • Vol. XIV - n° 4 - avril 2010
31 avaient entre 70 et 79 ans et 8, plus de 80 ans. Sur les 39 patients, 27 (69 %) restaient
oculaires pures (grade I) sur toute la durée de leur suivi, 12 (31 %) ont progressé vers une
myasthénie généralisée (grades II et III). Il n’y avait aucune différence significative entre les
deux groupes quant à l’âge, le sexe et le temps de suivi. Parmi les patients avec myasthénie
secondairement généralisée, aucun n’a eu besoin de ventilation assistée ou d’alimentation
par tube. Il en est de même du taux des anticorps anti-AChR (pour tout le groupe: 89 %
de positifs), mais pas de différences entre les deux groupes. Les tests électrophysiologiques
n’ont pas montré non plus de différence significative entre les deux groupes. Les affections
auto-immunes associées ou la présence de thymome ne sont pas non plus significativement
différentes. En revanche, le traitement immunomodulateur semble protéger les patients
d’une généralisation de la myasthénie, ce qui paraît constituer ainsi la seule leçon pratique
que l’on peut tirer de cette étude.
P.B.
Une myopathie immunitaire méconnue et rare,
mais traitable : la “SLONM”
Les myopathies à bâtonnets (myopathie némaline) sont réparties en deux grands groupes: les
formes congénitales héréditaires, qui donnent des tableaux parfois très sévères à la naissance,
ou parfois plus bénins, diagnostiqués à l’âge adulte (mais toujours avec un temps initial dans
l’enfance et l’adolescence), et les formes acquises, qui surviennent à l’âge adulte et qui sont
beaucoup plus évolutives. Des formes ont ainsi été décrites dans le cadre d’une séropositivité au
VIH. En dehors de ce cadre, les myopathies à bâtonnets sporadiques tardives (ou Sporadic Late
OnsetNemaline Myopathies [SLONM]) sont des atteintes déficitaires motrices subaiguës, souvent
assez évolutives et touchant particulièrement les membres supérieurs, le tronc et les muscles du
cou (bras ballants, tête tombante, etc.). Le diagnostic suspecté est souvent une polymyosite, mais
les CPK sont normales ou peu augmentées, il n’y a pas sur la biopsie musculaire les signes habi-
tuels de nécrose et surtout d’inflammation, et pas d’amélioration sous corticoïdes. Le diagnostic
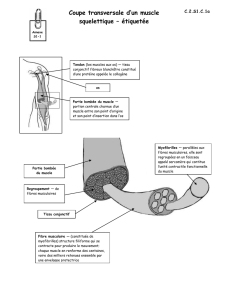
repose sur la mise en évidence de bâtonnets dans des fibres atrophiques (figure), présence
confirmée en microscopie électronique. Parfois, ces bâtonnets sont difficiles à mettre en évidence
et peuvent ne pas être repérés sur une première biopsie musculaire. Il existe souvent, associée à
ces cas, une gammapathie monoclonale (IgG ou IgA) sans hémopathie maligne. Ces formes avec
gammapathie monoclonale sont d’évolution particulièrement défavorable malgré les corticoïdes
et autres immunosuppresseurs associés. Des cas, ayant fait l’objet de publications récentes, ont
pu être stabilisés, voire améliorés sous autogreffe de cellules souches. Les auteurs de cet article
rapportent deux nouveaux cas associés à des IgG monoclonales, de signification indéterminée :
2 hommes, âgés de 61 et 46 ans, et présentant une forme sévère avec atteinte proximale, axiale
(tête tombante) et diplégie faciale. Ces 2 patients ont montré une nette amélioration sous Ig i.v.:
pour le premier patient, ce traitement seul a été
instauré, mais avec la nécessité de perfusions
tous les mois y compris 3 à 4 ans après le début,
avec uniquement la possibilité de réduire la dose
par cures de 50 g; le second patient a été placé
sous Igi.v. associée à des bolus de méthylpred-
nisolone puis à du mycofénolate mofétil (MMF)
jusqu’à une réduction des perfusions tous les
2 mois. La poursuite de l’amélioration après Ig
i.v. semblait se maintenir à distance de l’instau-
ration du traitement malgré cette dépendance.
T. Maisonobe,
hôpital de la Pitié-Salpêtrière, Paris
Commentaire
Il est intéressant de constater une thérapeutique
active immunitaire sous la forme d’Ig i.v. dans cette
affection musculaire acquise sévère et réputée
d’évolution très défavorable, allant souvent jusqu’à
l’atteinte respiratoire et la nécessité d’une ventila-
tion assistée. Cette expérience ne correspond pas
à l’impression des équipes de myologistes améri-
caines, françaises et belges qui ont déjà publié à
propos de cette pathologie et qui décrivaient au
contraire une résistance à toutes thérapeutiques
immunitaires ou alors seulement une sensibilité à
des chimiothérapies lourdes – de type melphalan
– ou à des greffes de moelle. Il y a trop peu de cas
traités par Ig i.v. pour que l’on puisse réellement en
tirer une conduite thérapeutique claire. Cependant,
l’amélioration de ces 2 patients, qui semble réelle
sur l’évolution des scores RMC, est un argument
supplémentaire quant à l’origine immunitaire (plus
ou moins liée à la gammapathie) de cette myopathie
et incite à essayer 2 ou 3 cures d’Ig i.v. systématiques
en cas de diagnostic avant de passer à des traite-
ments hématologiques plus lourds en cas d’échec.
Référence bibliographique
Milone M, Katz A, Amato A et al. Sporadic late onset nema-
line myopathy responsive to Igi.v. and immunotherapy.
Muscle Nerve 2010;41:272-6.
Figure. Muscle transversal, congelé, coloration
par le trichrome de Gomori modifié. Présence
dans une fibre atrophique d’images fuscino-
philes allongées évocatrices de bâtonnets.
Commentaire
Cette étude apporte des résultats intéressants
concernant les patients seniors avec pure myasthénie
oculaire, dont 31 % ont développé par la suite une
forme généralisée, sans qu’aucun caractère au début
permette de prédire la généralisation. Il faut souli-
gner que le traitement immunomodulateur serait
un bon moyen de prévenir la généralisation.
Les formes oculaires pures du sujet âgé sont
fréquentes et sont généralement de pronostic rela-
tivement bon, à condition de traiter ces patients.
Référence bibliographique
Allen JA, Scala S, Jones HR. Ocular myasthenia gravis in
a senior population: diagnosis, therapy, and prognosis.
Muscle Nerve 2010;41:379-84.

REVUE DE PRESSE
La Lettre du Neurologue Nerf & Muscle • Vol. XIV - n° 4 - avril 2010 | 125
Un nouveau phénotype de SLA familiale
avec mutation SOD1 (A4V)
La sclérose latérale amyotrophique (SLA) peut être familiale dans 10 à 15 % des cas. Les
mutations sur le gène codant pour l’enzyme superoxyde dismutase cytosolique Cu/Zn
(SOD1) expliquent 20 à 25 % de ces formes familiales, dont la plus commune montre
une substitution d’une alanine par une valine au codon 4 (A4V). Cette mutation concerne
jusqu’à 50 % des formes familiales du Nord des États-Unis. Son phénotype est caractérisé
par un mode de début et une évolution rapides, ainsi que par une survie moyenne située
entre 0,9 et 1,4 an. Le site de début est le plus souvent aux membres inférieurs, mais il peut
être bulbaire. L’atteinte du motoneurone spinal est en général prédominante, et l’atteinte
centrale n’est trouvée que dans 10 % des séries. Une hyperacousie et une ophtalmoplégie
sont rarement rapportées.
Cet article fait état du cas clinique et électrophysiologique d’un homme canadien âgé de
73 ans ayant 2 antécédents familiaux de SLA (grand-père maternel et cousin germain).
Le premier symptôme de ce patient est une paralysie faciale unilatérale qui se bilatéralise
en quelques semaines, avec dysarthrie et dysphagie. La paralysie d’une corde vocale est
constatée en fibroscopie. La diplégie faciale est massive à 4 mois du début de la maladie,
accompagnée d’un déficit moteur proximal de 2/5 à la main droite et de 4/5 à la cuisse
droite également. Les fasciculations sont notées mais peu abondantes ; les réflexes sont
diminués aux membres supérieurs, et sont normaux aux membres inférieurs. Une atteinte
diaphragmatique s’installe rapidement et, malgré une trachéotomie, le patient décède
moins de 15 mois après le début des signes.
N. Le Forestier,
hôpital de la Pitié-Salpêtrière, Paris
Commentaire
En dehors de l’intérêt de ce perturbant nouveau
phénotype, les auteurs comparent ce cas aux
formes familiales de la maladie du motoneu-
rone associées à une mutation de la sous-unité
p150Glued de la dynactine, protéine entrant
dans la constitution des microtubules moteurs et
essentielle pour le transport rétrograde axonal.
La forme clinique commence par une paralysie
précoce des cordes vocales, associée plus tardive-
ment à un déficit des muscles de la face, des mains
et de la partie distale des membres inférieurs. Les
auteurs repportent également les cas de maladie
du motoneurone de Madras et de syndrome de
Brown-Vialetto-Van Laere qui peuvent se présenter
par une atteinte des nerfs crâniens, une perte audi-
tive, une atteinte respiratoire et un déficit moteur
des membres inférieurs.
Ce court article est à connaître. Dommage, simple-
ment, que rien ne soit dit des deux cas familiaux
antérieurs de ce patient.
Référence bibliographique
Salameh JS, Atassi N, David WS. SOD1 (A4V)-mediated
ALS presenting with lower motor neuron facial diplegia
and unilateral vocal cord paralysis. Muscle Nerve
2009;40:880-2.
Abonnez-vous en ligne !
Bulletin d’abonnement
disponible page 143
www.edimark.fr
1
/
3
100%