>

5
La Lettre du Neurologue - Suppl. Les Actualités au vol. X - n° 5 - mai 2006
ACTUALITÉS
neurosciences
neurosciences
>
ACTUALITÉS
neurosciences
neurosciences
> Behavioural Brain Research
> European Journal
of Neuroscience
> Nature
> NeuroImage
> Neuron
> Molecular psychiatry
>Science
> Trends in Neuroscience
> Neurobiology of Learning
and Memory
> PNAS
L
a maladie de Huntington est une
maladie neurodégénérative hérédi-
taire incurable qui se manifeste par des
troubles moteurs de type mouvements
choréiques, des troubles cognitifs et
psychiatriques. Les symptômes se décla-
rent généralement entre 35 et 42 ans et
le décès survient après 15 à 20 ans
d’évolution. Six mille personnes en
France sont atteintes de la maladie de
Huntington, et plus de 12 000 porteurs
de la mutation sont provisoirement
indemnes de tout signe clinique. L’ori-
gine de cette maladie fut découverte en
1993, grâce à l’étude de familles véné-
zuéliennes particulièrement touchées
par la maladie de Huntington. Il s’agit
d’une maladie génétique autosomale,
liée à une expansion anormale de tri-
plets CAG dans l’exon 1 du gène IT15,
qui se traduit par une extension de glu-
tamines (poly-Q) dans la partie N-ter-
minale de la protéine huntingtine. La
huntingtine est une grosse protéine
dont le rôle précis demeure mal connu
mais qui possède de nombreux parte-
naires intracellulaires. Elle semble être
impliquée dans de multiples fonctions
intracellulaires, dont la régulation de la
transcription, le trafic et l’assemblage
protéiques. Elle joue un rôle important
dans la survie cellulaire, puisque des
souris génétiquement modifiées, qui
n’expriment plus la huntingtine, meu-
rent précocement à l’âge embryonnaire.
L’origine moléculaire de la maladie de
Huntington pourrait alors reposer sur
une perte de fonction vitale de la hun-
tingtine normale. Elle peut également
impliquer un gain de fonction toxique
de la huntingtine mutée, puisque la
présence d’une répétition de plus de
35 glutamines en N-terminal confère à
la protéine de nouvelles propriétés,
dont celle de former des agrégats intra-
neuronaux, et la rend toxique pour les
neurones.
U
ne caractéristique importante de la
maladie de Huntington est la vulné-
rabilité particulière d’une région céré-
brale donnée : le noyau caudé-putamen
(autrement appelé striatum). Ainsi, bien
que la huntingtine soit exprimée de
façon ubiquitaire, aussi bien dans le sys-
tème nerveux central que dans les tissus
périphériques, seuls les neurones du
striatum dégénèrent dans la maladie de
Huntington, tout au moins aux stades les
plus précoces de la maladie, et leur dys-
À la découverte
d’un double rôle de la dopamine
dans la maladie de Huntington
D. Charvin*, J. Caboche*
>
Charvin D, Vanhoutte P, Pages C et al. Unraveling a role for dopamine in Huntington’s disease:
the dual role of reactive oxygen species and D2 receptor stimulation. Proc Natl Acad Sci
2005;102(34):12218-23.
>
* UMR 7102, CNRS et université Paris-VI, Paris.
La maladie de Huntington :
principalement une maladie
du striatum
>

La Lettre du Neurologue - Suppl. Les Actualités au vol. X - n° 6 - juin 2006
ACTUALITÉS
neurosciences
neurosciences
>
6
> Behavioural Brain Research
> European Journal
of Neuroscience
> Nature
> NeuroImage
> Neuron
> Molecular psychiatry
>Science
> Trends in Neuroscience
> Neurobiology of Learning
and Memory
> PNAS
P
lusieurs données anatomiques et
expérimentales laissent penser que la
dopamine pourrait être impliquée dans la
maladie de Huntington. Par exemple, chez
les malades, la perte des neurones stria-
taux progresse selon un axe dorso-ventral
qui correspond au gradient de concentra-
tion de dopamine dans le striatum. D’autre
part, en 1998, l’équipe de J. Morton a
montré, dans un modèle murin pharmaco-
logique de la maladie de Huntington, que
la lésion des afférences dopaminergiques
protégeait les neurones striataux de la
mort. De plus, chez les souris knock-out
pour le transporteur de la dopamine, DAT,
qui présentent des concentrations de
dopamine anormalement élevées dans les
synapses, on observe une neurodégéné-
rescence striatale spontanée et des alté-
rations comportementales rappelant celles
de la maladie de Huntington, et qui se
déclarent spécifiquement au cours du
vieillissement des souris. Malgré l’exis-
tence de ces arguments indirects, il n’exis-
tait pas, jusqu’alors, de lien direct entre la
dopamine et la toxicité de la huntingtine
mutée dans les neurones striataux.
fonction constitue la base des symp-
tômes de cette maladie. L’origine de la
vulnérabilité préférentielle des neurones
striataux à la toxicité de la huntingtine
mutée demeure une question à laquelle il
semble difficile de trouver une réponse,
mais qui devrait permettre de trouver une
thérapie contre la maladie, à ce jour
incurable. Les neurones striataux pour-
raient comporter des éléments intrin-
sèques qui les rendent plus vulnérables
que les autres cellules à l’expression de la
huntingtine mutée. Par exemple, les neu-
rones striataux semblent souffrir d’un
déficit énergétique qui leur est propre
dans la maladie de Huntington. Ce sont
aussi les premières cellules dans les-
quelles se forment les agrégats de hun-
tingtine mutée. L’environnement de ces
cellules pourrait également contribuer à
cette situation. Ainsi, un défaut d’apport
du facteur neurotrophique BDNF (brain
derived neurotrophic factor) par les affé-
rences corticales pourrait être impliqué
dans la vulnérabilité des neurones stria-
taux, bien que ces cellules ne soient pas
les seules à recevoir du BDNF dans le sys-
tème nerveux central. En revanche, le
striatum est la structure cérébrale qui
reçoit la plus dense innervation dopami-
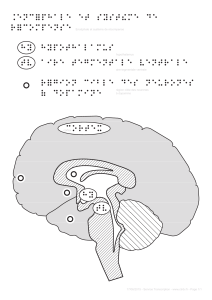
nergique. La dopamine est un neuro-
modulateur qui assure de multiples
fonctions physiologiques telles que le
contrôle de la coordination motrice, de
l’humeur, et les processus d’apprentissage
liés à la récompense. Toutefois, sous
certaines conditions, la dopamine peut
avoir un effet neurotoxique, aussi bien in
vitro qu’in vivo. C’est le cas par exemple
lorsque de trop fortes doses d’amphéta-
mine (un agoniste indirect de la dopa-
mine) sont administrées chez l’animal.
Nous avons donc envisagé la possibi-
lité que la vulnérabilité spécifique des
neurones striataux dans le cadre de la
maladie de Huntington puisse être liée
à leur innervation massive par les voies
dopaminergiques.
téine, soit 103 glutamines pour sa forme
mutée. Ces séquences sont fusionnées à
leur extrémité 3’ avec celle codant la
green fluorescent protein (GFP), ce qui
permet d’identifier les neurones trans-
fectés par émission de fluorescence et
de suivre la localisation cellulaire de
la huntingtine. Nous avons choisi ces
constructions pour plusieurs raisons :
– l’équipe de G. Bates a démontré en
1996 que la surexpression de l’exon 1 du
gène humain codant la huntingtine
mutée (avec 115 glutamines) est suffi-
sante pour reproduire un phénotype de
la maladie de Huntington chez la souris ;
– le fragment N-terminal codé par cette
construction possède les caractéristiques
importantes de la huntingtine mutée,
telles que la capacité de former des agré-
gats et la possibilité d’influencer les
interactions de la huntingtine avec
nombre de ses partenaires protéiques.
Nous avons démontré que ce modèle cel-
lulaire reproduisait deux caractéristiques
neuropathologiques importantes de la
maladie de Huntington, à savoir la for-
mation d’agrégats de huntingtine mutée
dans tous les compartiments intraneuro-
naux : noyau, soma, neurites dans les
neurones striataux, puis la dégénéres-
cence progressive de ces neurones, qui
se caractérise par la rétraction des neu-
rites précédant la condensation puis la
fragmentation du noyau. Tous les effets
observés dans ce modèle sont spéci-
fiques à la forme mutée de la huntingtine
puisque, par comparaison, l’expression du
fragment N-terminal de la huntingtine
normale, contenant 25 glutamines, ne
forme pas d’agrégat et n’est pas toxique
pour les neurones striataux. Nous avons
appliqué sur ce modèle cellulaire des
doses de dopamine subtoxiques, c’est-
à-dire induisant peu d’effet par elles-
mêmes sur la mort striatale, et analysé
les effets de ce traitement sur la toxi-
cité de la huntingtine mutée.
La dopamine,
suspectée de jouer un rôle
dans la maladie de Huntington
>
A
fin de déterminer de manière
directe si la dopamine a un rôle à
jouer dans la maladie de Huntington,
nous avons utilisé un modèle in vitro de
cultures primaires de neurones stria-
taux. Ceux-ci sont transfectés transitoi-
rement avec des ADN complémentaires
codant l’exon 1 du gène humain de la
huntingtine, possédant soit 25 gluta-
mines pour la forme normale de la pro-
À la recherche d’un rôle
éventuel de la dopamine
dans la maladie de Huntington
>

La Lettre du Neurologue - Suppl. Les Actualités au vol. X - n° 6 - juin 2006 7
mation d’agrégats protéiques inso-
lubles. Ces agrégats sont formés, dans
le cadre de la maladie de Huntington,
par des fragments N-terminaux de hun-
tingtine mutée, que reproduit notre
modèle cellulaire. L’implication directe
de ces agrégats dans la toxicité de la
huntingtine reste sujet à controverse.
Toutefois, leur présence témoigne
d’une souffrance cellulaire. Nous avons
démontré dans notre modèle que le
nombre de neurones transfectés pré-
sentant la huntingtine mutée sous
forme d’agrégats est fortement aug-
menté en présence de dopamine. Cet
effet, qui lui aussi est précoce, est
entièrement médié par l’activation des
récepteurs D2. En effet, la potentiali-
sation de la formation d’agrégats de
huntingtine mutée par la dopamine est
totalement mimée par un agoniste des
récepteurs D2 (le quinpirole), complè-
tement inhibée par un antagoniste des
récepteurs D2 (le raclopride), et n’est
pas reproduite dans les neurones stria-
taux provenant de souris knock-out
pour le récepteur D2.
D
ans notre modèle cellulaire, la dopa-
mine augmente la toxicité de la
huntingtine mutée dans les neurones
striataux, et ce via deux effets. Le pre-
mier repose sur la production de radi-
caux libres et l’activation de la voie pro-
apoptotique JNK/c-Jun. Le deuxième
implique l’activation des récepteurs D2.
En bloquant l’un ou l’autre de ces effets,
L
a voie JNK est une voie de MAP-
kinases qui est importante pour la
mise en place d’un programme proapop-
totique. Cette voie est notamment
impliquée dans la mort neuronale dans
plusieurs modèles de maladies neurodé-
génératives, telles que les maladies de
Parkinson et d’Alzheimer. Notre équipe a
montré, dans un premier temps, que
cette voie JNK est activée spécifique-
ment dans le striatum dans un modèle
pharmacologique in vivo de la maladie
de Huntington. In vitro, sur les cultures
primaires de neurones striataux, la hun-
tingtine mutée est capable d’activer JNK
et sa cible nucléaire, le facteur de trans-
cription c-Jun, dans une population
relativement faible de neurones. Nous
avons montré, dans notre modèle in
vitro, que la dopamine, en produisant
des radicaux libres, exerce un effet
synergique avec la huntingtine mutée
sur l’activation de la voie JNK/c-Jun
dans les neurones striataux. Ce méca-
nisme est précoce dans la pathophysio-
logie induite par la huntingtine mutée,
puisqu’il apparaît avant la formation des
agrégats nucléaires et avant toute souf-
france neuronale.
une partie significative des effets de la
dopamine sur la mort des neurones stria-
taux est inhibée. Ce n’est qu’en pré-
sence d’un cotraitement, bloquant à la
fois la production de radicaux libres et
l’activation des récepteurs D2, que les
effets de la dopamine sont complète-
ment inhibés.
L
es résultats issus de notre étude
dévoilent pour la première fois les
mécanismes moléculaires qui impli-
quent la dopamine dans la vulnéra-
bilité des neurones striataux dans le
cadre de la maladie de Huntington.
Cette implication est double, mettant
en jeu la production de radicaux libres
par la dopamine et l’activation des
récepteurs D2. L’auto-oxydation de la
dopamine, source de radicaux libres,
augmente dans le striatum au cours du
vieillissement. De ce fait, la dopamine
pourrait renforcer fortement, dans les
neurones striataux, l’activation de la
voie proapoptotique JNK, qui peut être
faiblement stimulée par la huntingtine
mutée seule, et contribuer ainsi à la
vulnérabilité préférentielle de ces cel-
lules dans la maladie de Huntington.
Cela pourrait en outre expliquer l’appa-
rition tardive des symptômes malgré
l’expression de la huntingtine mutée
depuis le développement embryon-
naire. La formation d’agrégats de hun-
tingtine mutée sous activation des
récepteurs D2 témoigne d’une souf-
france accrue des neurones porteurs de
ce type de récepteurs, qui sont préci-
sément, au sein du striatum, ceux qui
dégénèrent les premiers chez les
malades. De plus, les régions cérébrales
dans lesquelles ces neurones projettent
leurs axones – en particulier le globus
En produisant des radicaux
libres, la dopamine agit en
synergie avec la huntingtine
mutée pour activer la voie
proapoptotique de JNK/c-Jun
>
La dopamine augmente
la toxicité de la huntingtine
mutée dans les neurones
striataux, à la fois par
la production de radicaux
libres et par l’activation
des récepteurs D2
>
Perspectives
et applications thérapeutiques
>
C
omme dans de nombreuses mala-
dies neurodégénératives, l’une des
caractéristiques neuropathologiques
de la maladie de Huntington est la for-
La dopamine augmente
la formation d’agrégats
de huntingtine mutée,
via l’activation des récepteurs
dopaminergiques de type D2
>

La Lettre du Neurologue - Suppl. Les Actualités au vol. X - n° 6 - juin 2006
ACTUALITÉS
neurosciences
neurosciences
>
8
> Behavioural Brain Research
> European Journal
of Neuroscience
> Nature
> NeuroImage
> Neuron
> Molecular psychiatry
>Science
> Trends in Neuroscience
> Neurobiology of Learning
and Memory
> PNAS
neurones striataux exprimant la hun-
tingtine mutée et recevant de la dopa-
mine, in vitro. Le traitement avec un anti-
oxydant, l’ascorbate, s’est déjà montré
bénéfique sur les symptômes moteurs
de souris transgéniques modèles de la
maladie de Huntington. L’inhibition de
la voie JNK/c-Jun est quant à elle une
stratégie thérapeutique de plus en plus
étudiée pour soigner les maladies neuro-
dégénératives, telles que la maladie de
Parkinson. L’autre stratégie thérapeutique
identifiée dans notre étude consisterait
à bloquer l’activation des récepteurs
dopaminergiques de type D2 à un stade
pallidus externe – sont précisément
celles dans lesquelles les agrégats neu-
ritiques apparaissent en premier. Nos
résultats permettent donc de concilier
ces observations neuro-anatomiques et
pathophysiologiques.
Notre étude a permis d’identifier plu-
sieurs cibles thérapeutiques promet-
teuses pour la maladie de Huntington.
L’une de ces cibles est la production de
radicaux libres par la dopamine, ou l’acti-
vation de la voie JNK/c-Jun. En effet,
le blocage de l’un ou l’autre de ces évé-
nements est neuroprotecteur pour les
précoce de la maladie. Cela pourrait être
réalisé par un traitement aux neuro-
leptiques dès les stades présympto-
matiques. Ce type de traitement est
largement utilisé dans le cadre de la
schizophrénie, et pourrait permettre de
ralentir la progression de la maladie de
Huntington.
Ces différentes pistes d’investigation
sont d’autant plus intéressantes qu’il
n’existe à l’heure actuelle aucun traite-
ment permettant de ralentir la progres-
sion de la maladie, qui conduit inexora-
blement à la mort des patients.
■
1
/
4
100%