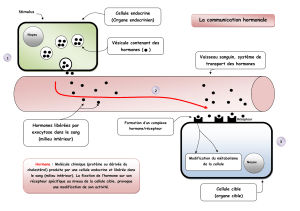
Dossier Pegvisomant, un antagoniste de l’hormone de croissance Antagonistes de la GH :

Dossier
Dossier
20
Métabolismes Hormones Diabètes et Nutrition (VII), no1, janvier/février 2003
Pegvisomant,
un antagoniste de l’hormone de croissance
V. Goffin*, P. Touraine**
Antagonistes de la GH :
quelles applications ?
L’acromégalie est une pathologie
résultant d’un excès de sécrétion
d’hormone de croissance (GH pour
growth hormone), le plus générale-
ment causée par un adénome hypo-
physaire affectant les cellules somato-
tropes. Les manifestations cliniques
de l’acromégalie sont une croissance
excessive de nombreux tissus (tissus
mous, os des mains et des pieds, infil-
tration des tissus mous), des maux
de tête ou encore une altération
visuelle par phénomène compressif
de la tumeur hypophysaire, autant
de signes qui peuvent être associés
à des troubles cardiovasculaires, une
hypertension, une résistance à l’insu-
line ou encore à diverses formes de
tumeur. En l’absence de traitement
adéquat, l’acromégalie s’associe à
un taux de mortalité élevé, résultant
principalement de ses conséquences
cardiovasculaires et néoplasiques (1).
Les traitements actuels de cette patho-
logie relèvent de la chirurgie (adé-
nomectomie), de la radiothérapie ou
de l’hormonothérapie (administra-
tion de somatostatine/dopamine qui
diminue la sécrétion hypophysaire
de GH). Cependant, cette dernière
approche n’est pas toujours efficace
pour abaisser de manière satis-
faisante les taux de GH circulants
ni, en corollaire, ceux de son second
messager, l’IGF-1 (insulin-like
growth factor 1). De même, ils ne
* INSERM U584, cibles tissulaires et moléculaires
des hormones, faculté de médecine Necker.
** Département d'endocrinologie et médecine
de la reproduction, hôpital Necker-Enfants-
Malades, Paris.
▲
▲
L’acromégalie est une pathologie résultant d’un excès de sécrétion d’hor-
mone de croissance liée, dans la grande majorité des cas, à un adénome
hypophysaire (cellules somatotropes). Les manifestations cliniques sont
une croissance générale accrue (os longs, tissus) et diverses pathologies
associées (risque cardiovasculaire, cancers). Ces effets résultent de l’acti-
vation du récepteur somatogénique mais peuvent également impliquer le
récepteur de la prolactine, auquel la hGH est également capable de se lier.
▲
▲
Les thérapies actuelles impliquent essentiellement les approches classiques :
neurochirurgie (adénomectomie), radiothérapie et hormonothérapie
(analogues de somatostatine/dopamine). Bien que souvent satisfaisantes,
ces approches ne permettent pas toujours d’inhiber efficacement les effets
de l’hypersécrétion de GH (growth hormone).
▲
▲
Le pegvisomant est un antagoniste de la GH. D’un point de vue moléculaire,
il s’agit d’un variant de hGH contenant 9 mutations ponctuelles. Ses pro-
priétés antagonistes résultent d’une seule mutation (la glycine de l’hélice 3),
les 8 autres mutations lui conférant entre autres une spécificité de liaison
pour le seul récepteur somatogénique. Enfin, pour augmenter la demi-vie
du pegvisomant in vivo, ce variant est complexé à des molécules de poly-
éthylène glycol (PEG), ce qui diminue sa filtration rénale et contribue éga-
lement à diminuer son potentiel immunogène.
▲
▲
Le pegvisomant cible non pas la tumeur hypophysaire (c’est-à-dire la
réduction de la synthèse de GH) mais l’action biologique de la GH sur ces
tissus cibles. Il se lie au récepteur de la GH sans l’activer et, par un simple
phénomène de compétition avec la GH endogène, l’empêche d’exercer
ses effets. Il n’a donc théoriquement aucun effet biologique intrinsèque.
▲
▲
Contrairement aux analogues de somatostatine/dopamine, le pegvisomant
ne réduit pas les taux circulants de GH ni le volume de la tumeur hypo-
physaire ; parfois même, il a un effet inverse. Cependant, il bloque l’action
biologique de la GH, ce qui se traduit par la baisse des taux circulants
d’IGF-1 (second messager de la GH). L’efficacité du traitement s’apprécie
donc par la mesure des taux d’IGF-1 et non, comme dans les traitements
hormonaux classiques (octréotide, lanréotide), par la baisse des taux de GH.
▲
▲
Dans les études cliniques réalisées à ce jour, le pegvisomant permet de
normaliser les taux d’IGF-1 chez plus de 90 % des patients acromégales
traités quotidiennement (20 mg en s.c.), avec réduction des désordres
associés (gonflements des tissus mous, maux de tête, asthénie, etc.). Le
traitement est globalement bien toléré, avec cependant quelques cas
d’effets secondaires (croissance du volume tumoral, hépatotoxicité).
▲
▲
Quelques données expérimentales (modèles cellulaires et/ou animaux)
suggèrent que les antagonistes de la GH pourraient également avoir un
effet bénéfique dans d’autres pathologies, comme la croissance de certains
cancers, la néphropathie ou la rétinopathie diabétique. Les données
cliniques sont cependant encore à venir.
▲
▲
Le pegvisomant n’est pas encore disponible sur le marché français.
points FORTS

Métabolismes Hormones Diabètes et Nutrition (VII), no1, janvier/février 2003
21
permettent pas toujours de réduire
le volume de la tumeur hypophysaire.
La recherche d’autres stratégies
thérapeutiques reste donc d’actua-
lité. Parmi celles-ci, le pegvisomant,
un antagoniste de la GH, a fait une
entrée remarquée lors du 81econgrès
annuel de l’Endocrine Society en
1999 (San Diego, Californie) et est
actuellement en phase III d’essai
clinique. Si l’acromégalie est claire-
ment la pathologie la plus directe-
ment liée à un excès de sécrétion de
GH et constitue donc la première
cible thérapeutique du pegvisomant,
l’inhibition des actions de la GH
pourrait se révéler bénéfique pour
d’autres pathologies dans lesquelles
l’implication de cette hormone (ou
de l’IGF-1) semble progressivement
émerger, par exemple la néphro-
pathie diabétique, la rétinopathie
diabétique, le diabète non insulino-
dépendant ou encore certaines formes
de cancer. Les données relatives
aux effets du pegvisomant dans le
contexte de ces diverses pathologies
n’étant encore que très préliminaires,
nous ne nous y attarderons pas.
Dans le cadre de ce bref article de
synthèse, notre objectif est, d’une
part, de retracer les principales étapes
du développement des antagonistes
de la GH, dont la version thérapeu-
tique est le pegvisomant, d’autre
part, de résumer les quelques études
in vivo publiées au cours des deux
ou trois dernières années, qu’il
s’agisse de modèles expérimentaux
ou, pour les plus récentes, des essais
cliniques de phases I à III.
Aspects moléculaires
Historique
L’histoire du pegvisomant com-
mence dans la seconde moitié des
années 1980, lorsqu’un groupe amé-
ricain (Monsanto Corp., Saint Louis)
parvient à déterminer la structure
tridimensionnelle (3D) de l’hormone
de croissance porcine par cristallo-
graphie aux rayons X (2). Confirmant
les études antérieures de structure
secondaire, effectuées notamment
par dichroïsme circulaire, l’étude
cristallographique démontre la pré-
sence de quelque 50 % de structures
hélicoïdales, réparties en 4 hélices α
antiparallèles (figure 1a). Si, globa-
lement, cette structure tertiaire fai-
sait déjà partie des modes de replie-
ment connus pour d’autres protéines
(par exemple, le cytochrome c), la
structure de la GH apparaît cepen-
dant singulière dans la connectivité
des hélices, c’est-à-dire la disposition
des boucles reliant les hélices entre
Figure 1. De la structure tridimensionnelle de la GH aux souris naines.
a. Structure tridimensionnelle de la GH, composée de quatre hélices
α
antiparallèles. Les
numéros des hélices sont placés à l’extrémité N-terminale de chacune d’elles.
b. Représentation schématique d’un tonneau de 4 hélices amphiphiles parfaites. Les faces
hydrophobes des hélices (bleues) sont orientées vers l’intérieur de la protéine, alors que les
faces hydrophiles (oranges) pointent vers l’extérieur.
c.Projection de l’hélice 3 de la GH bovine (bGH) selon son axe longitudinal. L’hélice présente un
caractère amphiphile marqué bien qu’imparfait en trois positions (Glu 117, Gly 119 et Ala 122).
Un profil d’hydrophobicité parfait est obtenu lorsque ces trois résidus sont remplacés par des
acides aminés à caractère d’hydrophobicité/hydrophilicité approprié, générant le mutant de
bGH appelé M8.
d.Alors que les souris transgéniques exprimant la bGH ont un phénotype de gigantisme, celles
exprimant le mutant bGH-M8 ont un phénotype nain, indiquant que ce variant exerce une
activité antagoniste de la GH endogène.
Dossier
Dossier
hydrophile
hydrophobe
Ala122→Asp
Gly119→Arg
Gly117→Leu
A.
C.
D.
B.
1N
Ala
122 Asp
115 Glu
126Gly
119
Lys
112
Lys
114
Arg
125
Glu
118
Glu
111
Leu
123
Leu
116
Val
109
Ile
120
Leu
113
Met
124
Glu
117
Tyr
110
Leu
121
Asp
122 Asp
115 Glu
126Arg
119
Lys
112
Lys
114
Arg
125
Glu
118
Glu
111
Leu
123
Leu
116
Val
109
Ile
120
Leu
113
Met
124
Leu
117
Tyr
110
Leu
121
C
bGH
109-126 bGH M8
109-126
Transgénique
bGH-M8 Transgénique
bGH
sauvage
2
3
4

Dossier
Dossier
Métabolismes Hormones Diabètes et Nutrition (VII), no1, janvier/février 2003
22
elles (2). Cette connectivité particu-
lière fut d’ailleurs un des arguments
majeurs pour rassembler au sein
de la même famille des “cytokines
hématopoïétiques” des ligands aussi
différents que la prolactine et la
GH, l’érythropoïétine, la plupart
des interleukines ou encore la lep-
tine (3). Les spécialistes de l’ana-
lyse structurale des protéines ont
constaté que, dans ce type de struc-
ture 3D, les hélices présentent sou-
vent un caractère amphiphile (ou
amphipathique). En d’autres termes,
si l’on regarde ces hélices comme
des cylindres selon leur axe longi-
tudinal (figure 1b),la surface du
cylindre (c’est-à-dire de l’hélice)
orientée vers l’intérieur de la molé-
cule est généralement constituée
d’acides aminés à caractère globa-
lement hydrophobe, alors que la
moitié orientée vers l’extérieur est
constituée d’acides aminés à carac-
tère majoritairement hydrophile.
Cette distribution spécifique des
acides aminés au sein des hélices
dites “amphipathiques” n’est pas
fortuite, puisqu’elle contribue entre
autres à stabiliser la protéine, les
acides aminés hydrophobes des
faces internes des hélices α,créant
conjointement un “cœur hydro-
phobe” dont les molécules d’eau
sont exclues, alors que, au contraire,
l’hydrophilicité des faces externes
des hélices contribue à la solubilité
de la protéine dans les solvants
aqueux. Dans les GH porcines et
bovines, très similaires quant à leur
séquence, c’est surtout l’hélice 3,
comprenant les acides aminés 109 à
126, qui présente un caractère
amphiphile (figure 1c). Cette amphi-
philicité n’est cependant pas opti-
male, puisque la glycine 119, acide
aminé neutre en termes d’hydro-
philicité, et l’alanine 122 sont sur
la face exposée au solvant, alors que
le glutamate 117, un acide aminé
hydrophile et chargé négativement,
pointe, quant à lui, vers le cœur
hydrophobe de la protéine.
À la charnière des années 1980-1990,
le Dr J.J. Kopchick et ses collègues
(Ohio University) entreprennent de
générer un variant de GH dans
lequel l’amphiphilicité de l’hélice 3
allait être rendue “théoriquement”
parfaite, le but avoué étant d’obtenir
des superagonistes de la GH en vue
d’applications potentielles dans
des contextes pathologiques de
déficience en GH (4). Pour ce faire,
cette équipe remplace le glutamate
117 par une leucine (hydrophobe),
la glycine 119 et l’alanine 122 res-
pectivement par une arginine et un
aspartate, deux résidus chargés et
hydrophiles (figure 1c). Maîtrisant
parfaitement les techniques de
transgenèse, cette équipe produit
dans la foulée plusieurs lignées de
souris génétiquement modifiées,
exprimant ce variant de GH conte-
nant les 3 mutations ponctuelles sus-
mentionnées et dénommé bGH-M8.
Les effets de la surexpression de la
GH in vivo présentant l’immense
avantage d’être très rapidement
repérables par un phénotype de
gigantisme (figure 1d) d’installation
progressive dès la première quin-
zaine de vie postnatale, la seule
analyse visuelle de ces souris trans-
géniques allait permettre de se faire
une idée assez précise de l’activité
du mutant de GH surexprimé. Et là,
quelle ne fut pas la surprise de
constater que les souris exprimant le
mutant bGH-M8 présentaient un
phénotype de nanisme (figure 1d),
leurs taille et poids étant réduits de
manière proportionnelle au taux
d’expression de la protéine trans-
génique dans chaque souris analysée
(4) ! Alors que l’affinité du mutant
bGH-M8 pour le récepteur somato-
génique se révèle identique à celle
de l’hormone sauvage, ces observa-
tions montraient, pour la première
fois, que la liaison au récepteur et
la capacité à l’activer n’étaient pas
nécessairement deux événements
couplés, les auteurs proposant que
leur variant de bGH agisse in vivo
comme un antagoniste de l’hormone
endogène. L’obtention de lignées
transgéniques exprimant les variants
de GH contenant individuellement
l’une des trois mutations précitées
allait permettre d’identifier la seule
mutation de la glycine 119 comme
responsable du phénotype nain,
c’est-à-dire nécessaire et suffisante
pour générer un antagoniste de la GH
(5). Bien inspiré, le Dr Kopchick
protège ces mutants de GH par un
brevet (6) et publie ses résultats peu
après, se limitant à l’époque à
conclure que “l’hélice 3 est impor-
tante pour l’activité biologique de
la GH”. Sans le savoir encore,
l’histoire du pegvisomant venait de
commencer et, comme on l’aura
perçu, sur une observation totale-
ment fortuite.
Bases moléculaires
de l’activité antagoniste
des mutants de GH
À peu près à la même époque que
ces études pionnières menées à
l’Université d’Ohio, la société de
biotechnologie Genentech (South
San Francisco, Californie) lançait
une étude de grande envergure de
l’hormone de croissance humaine
(hGH), dont l’ADN complémen-
taire avait été cloné quelque dix ans
plus tôt par une équipe de l’UCSF
(7, 8). Cette analyse plurielle de la
hGH, focalisée sur des éléments
structuraux, biochimiques et bio-
logiques (9),allait amener des
connaissances essentielles dans la
compréhension des mécanismes
moléculaires régissant l’interaction
entre l’hormone et son récepteur
(GHR). Parmi les travaux les plus
marquants de cette vaste étude, la
détermination de la structure 3D du
complexe entre la GH et le domaine
extracellulaire du GHR (10) allait
rapidement devenir une donnée
incontournable dans le contexte de
l’activation des récepteurs de cyto-
kines (figure 2a). Ce fut en effet la
première démonstration claire de
l’hormone de croissance induisant
la dimérisation de son récepteur :
une seule molécule de GH interagit
avec deux molécules (identiques) de
récepteur (figures 2a et 3a). Cette
interaction implique deux régions
distinctes sur l’hormone, appelées
site de liaison 1 (entre GH et pre-

Dossier
Dossier
Métabolismes Hormones Diabètes et Nutrition (VII), no1, janvier/février 2003
23
mière molécule de GHR) et site de
liaison 2 (entre GH et seconde molé-
cule de GHR). Complémentant cette
analyse structurale pionnière, un
ensemble d’éléments biochimiques
allaient ensuite renforcer la caracté-
risation de cette interaction hormone-
récepteur. En 1992, le groupe du
Genentech publie dans la revue
Science l’article faisant désormais
référence pour les antagonistes de la
GH humaine (11). Comme l’on
pouvait s’y attendre à la lumière des
travaux antérieurs de Kopchick et
al. sur la GH bovine, le remplace-
ment de la glycine de l’hélice 3 de
la hGH par une arginine (mutant
G120→R) génère également un
antagoniste. Grâce à l’analyse de la
structure 3D du complexe GH/GHR
(10),le mécanisme sous-tendant les
propriétés antagonistes des mutants
de la glycine allait enfin être
décrypté. L’interaction entre le site
2 de l’hormone – auquel appartient
la glycine de l’hélice 3 – et le récep-
teur est directement corrélée à la
petite taille de cet acide aminé. En
effet, grâce à une chaîne latérale
réduite à sa plus simple expression
(un atome d’hydrogène), la glycine
contribue à l’existence d’une cavité
(figure 2b, droite) entre les hélices
1 et 3, dans laquelle viennent s’an-
crer deux résidus tryptophane du
récepteur (10). Le remplacement de
la glycine par quelque acide aminé
que ce soit, c’est-à-dire de l’hydro-
gène par toute autre chaîne latérale,
vient encombrer la cavité (figure 2b,
gauche),empêchant par là même
les résidus tryptophane du récepteur
d’établir une interaction fonction-
nelle avec le site 2 de l’hormone. En
corollaire, le variant G120R-hGH
(ou bGH-G119R) n’est plus capable
d’induire une dimérisation fonc-
tionnelle du GHR (figure 3b). En
revanche, comme son site de liaison 1
n’est pas altéré, il garde la capacité
de se lier au récepteur et peut donc
agir comme compétiteur de l’hor-
mone sauvage lorsque les deux
ligands sont mis en présence l’un de
l’autre (figure 3c),comme dans le
cas des souris transgéniques évoqué
plus haut (GH endogène et antago-
niste transgénique) (4). Le mutant
exerce alors une activité antagoniste
de manière dose-dépendante, par un
simple mécanisme de compétition
pour la liaison au récepteur. En
conclusion, et c’est un point extrême-
ment important à garder en mémoire
pour l’application clinique, les anta-
gonistes de la GH n’ont – jusqu’à
preuve du contraire – aucune acti-
vité intrinsèque, c’est-à-dire qu’ils
n’induisent aucun phénomène sur
les cellules cibles et n’agissent qu’en
empêchant la GH naturelle de se lier
à son récepteur et, ainsi, d’exercer
ses activités biologiques.
Développement du pegvisomant
Le mécanisme d’action des antago-
nistes de la GH contenant la mutation
“glycine” reposant sur une compéti-
tion avec la GH, cette propriété est
directement corrélée avec son affi-
nité de liaison pour le récepteur
somatogénique. La mutation de la
glycine n’affecte pas l’affinité de
liaison de l’hormone pour le GHR ;
lorsque les hormones sauvage et
mutée sont en quantité équimolaire,
on observe donc une diminution
d’activité de l’ordre de 50 %. En
corollaire, le mutant doit être pré-
sent en excès molaire par rapport
à la GH sauvage pour inhiber tota-
lement l’activité somatogénique. La
première étape du développement
d’un antagoniste utilisable dans
un contexte thérapeutique fut donc
d’améliorer l’affinité de liaison du
G120R-hGH. Grâce au criblage
d’envergure réalisé par phage dis-
play (analyse de l’effet de mutations
aléatoires sur l’affinité de la hGH
pour son récepteur soluble), plu-
sieurs substitutions d’acides aminés
ayant un effet bénéfique sur ce para-
mètre furent identifiées (12). Une
seconde version du G120R-hGH
répondant au nom de B2036 fut donc
générée, se différenciant du G120R-
hGH par la présence de 8 substitu-
tions additionnelles dans le site de
liaison 1 (12). De manière encore
mal comprise, cependant, l’augmen-
tation d’affinité (d’un facteur 5) de
ce nouveau variant, mise en évi-
dence sur le récepteur soluble, n’est
pas observée sur la forme membra-
naire du GHR (13),qui est et reste
la principale cible moléculaire des
antagonistes in vivo. Toutefois, le
B2036 présente un avantage non
négligeable sur le G120R-hGH. En
effet, la hGH a la capacité de se lier
non seulement au récepteur somato-
génique, mais également au récep-
teur lactogénique (ou récepteur de
la prolactine, le PRLR), très proche
du GHR d’un point de vue structural
(14). Comme la hGH naturelle, le
mutant G120R-hGH est capable de
se lier au PRLR et exerce également
des propriétés antagonistes sur ce
Figure 2. Bases structurales de l’action
antagoniste des mutants de la glycine de
l’hélice 3
a. Structure tridimensionnelle du complexe
hGH-hGHBP
2
(BP : domaine extracellulaire
du GHR). Cette structure cristallographique
fut la première démonstration claire de la
GH induisant la dimérisation de son récep-
teur, ce qui implique deux régions de l’hor-
mone (sites de liaison).
b. La glycine de l’hélice 3 permet l’existence
d’une cavité (trait blanc) entre les hélices 1
et 3. Lorsque la glycine est remplacée par
une arginine, cette cavité est occupée par la
chaîne latérale plus encombrante que celle
de la glycine (un hydrogène), ce qui crée un
encombrement stérique au sein du site de
liaison 2 de la GH (trait pointillé).
A.
B.
hGH
hGHBP1
hGHBP2
Hélice 3
Arginine Glycine

Dossier
Dossier
Métabolismes Hormones Diabètes et Nutrition (VII), no1, janvier/février 2003
24
récepteur in vitro (15). En revanche,
parmi les 8 mutations supplémen-
taires introduites dans le site 1 du
B2036, certaines affectent des rési-
dus clés de l’interaction hGH-PRLR
(16). C’est le cas notamment du
glutamate 174, résidu impliqué dans
la coordination d’un ion de zinc lors
de l’interaction hGH-hPRLR, et dont
la seule mutation en alanine diminue
350 fois l’affinité de la hGH pour le
récepteur soluble de la PRL (17).
En conséquence, ces 8 mutations
empêchent le B2036 de se lier au
récepteur de la PRL (18),ce qui
confère à ce mutant une spécificité
de liaison pour le seul GHR, avantage
non négligeable dans un contexte
thérapeutique, car il est toujours plus
complexe de contrôler les effets
d’une molécule ayant plusieurs cibles
moléculaires. Bien que le B2036
soit un antagoniste efficace de la
hGH, certaines observations expéri-
mentales suggèrent que ce variant
demeure capable d’induire la for-
mation de dimères de récepteurs,
sans qu’un tel dimère n’adopte tou-
tefois une conformation permettant
l’activation des voies de signalisa-
tion en aval (13). Cette observation
demande cependant confirmation.
Une dernière amélioration apportée
au cours du développement du peg-
visomant est le paramètre de sa
demi-vie in vivo. La GH (naturelle
ou mutée) a une demi-vie qui oscille
aux alentours de 15 à 60 minutes
selon l’espèce considérée (19).
Dans la perspective d’inhiber effi-
cacement et de manière prolongée
les effets biologiques résultant des
taux élevés de hGH, il est donc pré-
férable d’administrer un antagoniste
dont la demi-vie sera aussi longue
que possible. Dans cette optique,
des molécules de polyéthylène gly-
col (PEG5000) ont été greffées sur
l’antagoniste B2036, générant le
B2036-PEG ou pegvisomant (figure
2c, tableau I). La “PEGylation” a
plusieurs avantages. Le premier
concerne la taille apparente de la pro-
téine “PEGylée”. Une moyenne de
4 à 5 molécules de PEG5000 (masse
moléculaire de 5 000 daltons) étant
liées sur chaque protéine, la masse
moléculaire théorique du pegviso-
mant avoisine les 45-50 kDa, versus
22 kDa pour l’hormone “nue” (20).
Il est cependant probable que la
masse moléculaire apparente du
pegvisomant soit plus élevée encore,
l’encombrement stérique réel des
molécules de PEG étant, de par leur
structure polymérique, plus élevé
que celui d’une protéine globulaire
de masse équivalente. Il en résulte
une demi-vie nettement accrue (T1/2
de l’ordre de 15 heures), due essen-
tiellement à une diminution du taux
de filtration glomérulaire rénale
Figure 3. Bases moléculaires de l’action antagoniste du pegvisomant.
a.La GH active son récepteur en induisant sa dimérisation de manière séquentielle : le site de
liaison 1 d’abord, puis le site de liaison 2 ensuite interagissent chacun avec une molécule de
récepteur. Une fois dimérisé, le GHR active les cascades de signalisation intracellulaire, ce
qui conduit notamment à la synthèse de son second messager, l’IGF-1.
b. Dans le pegvisomant, le remplacement de la glycine de l’hélice 3 par une lysine génère un
encombrement stérique dans le site 2 (figure 2b),ne permettant plus à cette région d’établir
une liaison fonctionnelle avec le GHR. Ce mutant de hGH, bien qu’encore capable d’interagir
avec le récepteur via son seul site 1, est donc inactif, ce qui se traduit notamment par son
incapacité à induire l’expression d’IGF-1.
c. Lorsque le pegvisomant est mis en présence de la hGH naturelle, une compétition s’exerce
pour la liaison au GHR. Lorsque la concentration de pegvisomant est suffisante pour prendre
le pas sur la hGH endogène, l’effet antagoniste du mutant supplante l’effet agoniste de l’hor-
mone naturelle, ce qui se traduit par la baisse des taux circulants d’IGF-1.
PEG : polyéthylène-glycol.
A.
B.
C.
hGH
12
12 12
1
1
111
2
12
1
hGHR
hGH
inactif actif
actif inactif
membrane plasmique
inactif
IGF-1
membrane plasmique
membrane plasmique
Pegvisomant
PEG PEG
PEG
PEG PEG PEG
PEG
PEG
PEG PEG
PEG
PEG PEG
PEG
PEG PEG PEG
PEG
PEG
PEG PEG PEG
PEG
PEG
PEG
PEG
PEG
PEG
+ Pegvisomant
IGF-1
IGF-1
IGF-1
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%