Lire l'article complet

Médecine
& enfance
EXAMEN RÉDA
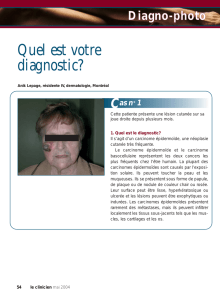
Le petit Réda, trois ans, originaire de Tunisie vient d’arriver en France. Vous le voyez pour la première fois et
constatez cette lésion du cuir chevelu (photo ci-dessous).Le reste de l’examen est sans particularité et le car-
net de vaccination est à jour. La maman vous dit que cette lésion est visible depuis la naissance.
Quel est votre diagnostic ?
COUP D’ŒIL
C. Guillaumat,
A. Mosca,
service de pédiatrie,
centre hospitalier
Sud francilien, Evry
Rubrique dirigée
par A. Mosca,
alexis.mosca@
ch-sud-francilien.fr
mai 2010
page 249
EXAMEN AMINATA
Aminata, huit jours, est adressée aux urgences pédiatriques par la PMI pour une tuméfaction axillaire gauche
découverte par la maman le matin même. Elle est née à terme, eutrophe, au terme d’un travail prolongé.
A l’examen, c’est un bébé tonique avec un comportement normal, qui boit bien. Elle présente une tumé-
faction axillaire gauche postérieure, de 1,5 cm de diamètre, d’aspect inflammatoire, mobile, dont la pal-
pation est dure et semble sensible (photo ci-dessous). Le reste de l’examen est sans particularité.
Quel est votre diagnostic ?
Voir les diagnostics page suivante ➝
129930 249-51 19/05/10 21:17 Page249

DIAGNOSTIC AMINATA :
CYTOSTÉATONÉCROSE
NÉONATALE
La cytostéatonécrose du nouveau-né
(CSN) est une hypodermite aiguë se dé-
veloppant durant les premiers jours de
vie. Elle survient chez un nouveau-né
en pleine santé, né à terme ou postma-
ture, au décours d’un accouchement
compliqué. Elle apparaît après un inter-
valle libre, dans les quinze à trente pre-
miers jours de vie.
Sa physiopathologie est encore mal
connue. Elle résulterait de l’association
de facteurs d’origine maternelle (hyper-
tension artérielle gravidique et pré-
éclampsie, diabète gestationnel, prise
d’inhibiteurs calciques ou de cocaïne
pendant la grossesse), de facteurs d’ori-
gine constitutionnelle (hypoxie péri-
phérique due à une anémie ou à une
thrombocytose néonatales), mais aussi
de facteurs obstétricaux (travail prolon-
gé, hypothermie, hypoxie, infection ma-
ternofœtale, utilisation d’outils conten-
dants).
La CSN débute par un érythème qui
évolue rapidement vers des placards
d’hypodermite rouge violine plus ou
moins diffus, douloureux dans 25 % des
cas. Cette inflammation s’atténue en
quelques semaines pour laisser place à
une atrophie du tissu sous-cutané qui
persistera plusieurs années, sans reten-
tissement fonctionnel particulier.
La CSN est le plus souvent localisée au
niveau du dos, du cou et des membres
supérieurs. Une souffrance néonatale
sévère s’accompagnera de lésions dif-
fuses, alors que les traumatismes locali-
sés, comme ceux dus à l’utilisation de
forceps, induisent en général des lé-
sions focales.
Le diagnostic est le plus souvent cli-
nique, mais, en cas de doute, il peut être
confirmé par une cytoponction qui trou-
ve des cellules multinucléées avec inclu-
sions vides correspondant à des cristaux
lipidiques.
L’hypercalcémie est la complication
la plus souvent rencontrée (30 à 75 %
des cas) et elle est plus fréquente dans
les formes disséminées. Elle est le plus
souvent asymptomatique mais impose
une surveillance régulière de la calcé-
mie (dosage hebdomadaire pendant
deux à trois mois). Elle peut apparaître
jusqu’à deux mois après le début des
signes cutanés et peut générer des dé-
pôts calciques viscéraux (néphroalcino-
se, lithiases rénales), le plus souvent
asymptomatiques et régressifs en
quelques mois.
Les complications locales sont domi-
nées par la douleur et l’atrophie sous-
cutanée.
Des hypertriglycéridémies majeures,
des thrombopénies sévères ou des cas
d’hypoglycémie ont été décrits pendant
la CSN. Le plus souvent asymptoma-
tiques, ces anomalies, qui peuvent ap-
paraître avant les signes cutanés, ré-
gressent avec la CSN.
Lors de la prise en charge d’un nou-
veau-né présentant une CSN, il est im-
portant de rassurer les parents sur la ré-
gression spontanée des lésions tout en
les prévenant des risques de dépression
cutanée sans conséquence esthétique
ou fonctionnelle. Le traitement de la
douleur, qu’il ne faut pas sous-estimer,
peut nécessiter l’utilisation de morphi-
niques. L’hypercalcémie doit être prise
en charge spécifiquement, et il est im-
portant de sensibiliser les parents aux
signes la faisant suspecter (vomisse-
ments, constipation, irritabilité, retard
de croissance). Dans les formes dissémi-
nées, il est recommandé de ne pas sup-
plémenter ces nourrissons par vitamine
D et d’interdire l’exposition solaire pen-
dant les premiers mois de vie.
DIAGNOSTIC RÉDA :
APLASIE CUTANÉE
CONGÉNITALE
L’aplasie cutanée congénitale corres-
pond à un défaut de développement du
tissu cutané, visible dès la naissance,
sur une surface de taille variable. C’est
une affection rare (0,03 % des nais-
sances), le plus souvent sporadique,
mais quelques cas familiaux à transmis-
sion autosomique dominante ont été
rapportés.
L’étiologie de l’aplasie cutanée
congénitale n’est pas univoque et diffé-
rents facteurs sont probablement intri-
qués dans sa pathogénie. Des facteurs
intra-utérins : rupture du tégument lors
de la forte croissance céphalique entre
la dixième et la dix-huitième semaine
de grossesse, traumatisme intra-utérin,
adhérences amniotiques, problème vas-
culaire (diabète maternel, épisodes
d’hypotension). On retrouve parfois un
facteur tératogène : antithyroïdiens de
synthèse, acide valproïque, IEC. Enfin,
une cause génétique est suspectée de-
vant des cas familiaux.
Cliniquement, on observe une absen-
ce de revêtement cutané d’une zone de
peau bien limitée. La localisation au
vertex, à proximité de la fontanelle pos-
térieure, est la plus fréquente (85 % des
cas). Elle est le plus souvent unique
(75 % des cas), mais il existe parfois des
lésions doubles (20 %), voire multiples
(5 %). En cas de localisation médiane,
on recherchera une anomalie de ferme-
ture pariétale. Plus rarement, cette lo-
calisation peut aussi s’associer à des
anomalies des extrémités (syndrome
d’Adams-Olivier) ou s’intégrer à un syn-
drome polymalformatif (syndrome de
Bart, de Johanson-Blizzard, syndrome
EEC, trisomie 13). Les lésions sont le
plus souvent superficielles et ne concer-
nent que l’épiderme, mais dans 15 à
20 % des cas le défaut de développe-
ment du cuir chevelu peut toucher la
voûte crânienne, voire la dure-mère.
Des complications infectieuses et hé-
morragiques sont alors à craindre, avec
une mortalité dans 20 à 55 % des cas.
Le diagnostic est clinique. L’IRM per-
met d’apprécier la profondeur de l’at-
teinte dans les formes étendues.
Il n’y a pas de consensus sur le traite-
ment. Les lésions superficielles limitées
à l’épiderme sont traitées par panse-
ments et antibiothérapie locale. Une
alopécie cicatricielle est alors constante.
En revanche, les formes étendues avec
atteinte du tissu sous-cutané nécessi-
Médecine
& enfance
mai 2010
page 250
129930 249-51 19/05/10 21:17 Page250

tent une chirurgie précoce pour éviter
les complications infectieuses et hémor-
ragiques. Il pourra s’agir de la réalisa-
tion de lambeaux de greffe cutanée, voi-
re de véritable chirurgie de reconstruc-
tion.
Médecine
& enfance
mai 2010
page 251
Pour en savoir plus
Sur la cytostéatonécrose néonatale :
MAHÉ E., DEPROST Y. : « La cytostéatonécrose du nouveau-
né »,
Ann. Dermatol. Vénéréol.,
2007 ;
134 :
494-8.
BURDE A.D., KRAFCHIK B.R. : « Subcutaneous fat necrosis of the
newborn : a review of 11 cases »,
Pédiatr. Dermatol.,
1999 ;
16 :
384-7.
MAHÉ E., GIRSZYN N. et al. : « Subcutaneous fat necrosis of the
newborn. A systematic evaluation of risk factors, clinical aspects,
complications and evolution in 16 children »,
Br. J. dermatol.,
2007 ; 156 : 709-15.
Sur l’aplasie cutanée congénitale :
ALOULOU H., CHAARI W., KHANFIR S. et al. : « Aplasia cutis
congenita du vertex (5 observations) »,
Arch. Pédiatr.,
2008 ;
15 :
382-7.
14eJournée de pathologie
infectieuse pédiatrique ambulatoire
Journée organisée par la revue Médecine et enfance
sous la direction scientifique de Robert Cohen
avec
Infovac-France
l’Association clinique et thérapeutique infantile du Val-de-Marne (ACTIV)
et le Groupe de pathologie infectieuse pédiatrique (GPIP)
Voir le programme page 215
BULLETIN D’INSCRIPTION
A LA 14eJOURNÉE DE PATHOLOGIE INFECTIEUSE PÉDIATRIQUE AMBULATOIRE
à envoyer à Médecine et enfance, 23 rue Saint-Ferdinand, 75017 Paris, accompagné de votre règlement
Frais de participation : 130 euros
NOM, Prénom
Adresse
Code postal, Ville
Courriel
Je joins un chèque de 130 euros à l’ordre de Médecine et enfance
Une confirmation d’inscription et un reçu vous seront envoyés par mail ou par courrier dès réception de votre bulletin d’inscription
☞
le samedi 9 octobre 2010
à la Maison de la Chimie
28 bis, rue Saint-Dominique, 75007 Paris
129930 249-51 19/05/10 21:17 Page251
1
/
3
100%