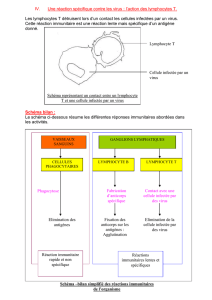
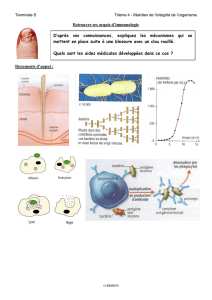
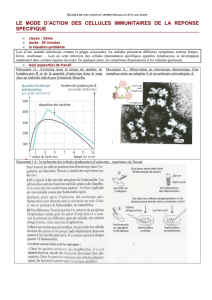
les mecanismes immunitaires de lutte contre l`infection hepatique

SYNTHESE BIBLIOGRAPHIQUE EN BIOLOGIE ET BIOTECHNOLOGIE
Mars 2013
LES MECANISMES IMMUNITAIRES DE LUTTE
CONTRE L’INFECTION HEPATIQUE
PAR LE VIRUS DE L’HEPATITE MURINE DE TYPE 3
Par
Marie-Charlotte Manus
MASTER 2 BIOLOGIE GESTION,
UNIVERSITE DE RENNES 1, UFR SCIENCES DE LA VIE ET DE L’ENVIRONNEMENT
Tuteur :
Claire Piquet-Pellorce

M.C. Manus / Les mécanismes immunitaires de lutte contre l’infection hépatique par le virus MHV3 ©
1
Remerciements
Je tiens à remercier Mme Claire Piquet-Pellorce, Maître de Conférences à l’Université de
Rennes1, pour son aide et ses conseils pour la rédaction de cette synthèse bibliographique.
«Le tuteur chercheur a pour rôle de conseiller l'étudiant, l'orienter dans ses recherches
bibliographiques, l'aider à comprendre les articles, en faire une synthèse de manière logique et
rigoureuse. Il ne peut vérifier toutes les citations et interprétations de l'étudiant. Il ne peut donc
s'engager vis à vis d'éventuelles erreurs ».

M.C. Manus / Les mécanismes immunitaires de lutte contre l’infection hépatique par le virus MHV3 ©
2
Les mécanismes immunitaires de lutte contre l’infection hépatique par le virus
de l’hépatite murine de type 3
M.C. Manus
Master Biologie, Gestion et Marketing – Université de Rennes I
Résumé
Le modèle de l’hépatite murine induite par le virus de l’hépatite murine de type 3 (MHV3)
permet d’étudier les causes d’une réponse inflammatoire anormale, ainsi que les mécanismes
immunitaires activés en réponse à l’infection. L’infection hépatique provoque rapidement le
recrutement et l’activation des cellules associées à la réponse immunitaire innée, les cellules
dendritiques, les cellules endothéliales sinusoïdales, les cellules de Kupffer, les cellules tueuses naturelles
(NK), les neutrophiles, ainsi que la sécrétion de chémokines et de cytokines pro-inflammatoires. La
rupture de l’état de tolérance hépatique accompagnée de la réponse inflammatoire permet l’activation
des cellules de l’immunité spécifique ou adaptative, les lymphocytes. La réponse immunitaire spécifique
de type Th1 confère aux souris infectées par le virus MHV3 une résistance à l’infection, contrairement à
la réponse immunitaire spécifique de type Th2.
Sommaire
Introduction ................................................................................................................................................... 3
I. Le virus de l’hépatite murine de type 3 : MHV3 ................................................................................... 4
A. Le virus MHV3 ................................................................................................................................... 4
B. Infection hépatique : Cellules cibles et variants de pathogénicité ................................................... 4
C. Les récepteurs des virus MHV ........................................................................................................... 5
II. Les mécanismes immunitaires face à l’infection hépatique par le virus MHV3 ................................... 6
A. Les mécanismes de l’immunité innée : réponse inflammatoire ....................................................... 6
B. Les mécanismes de l’immunité spécifique ou adaptative .............................................................. 12
Conclusions .................................................................................................................................................. 16
Références bibliographiques ....................................................................................................................... 17

M.C. Manus / Les mécanismes immunitaires de lutte contre l’infection hépatique par le virus MHV3 ©
3
Introduction
Les hépatites virales peuvent engendrer la mort des sujets contaminés, animaux ou humains, et
constituent donc un problème de santé majeur. En effet, une infection hépatique virale peut causer des
atteintes inflammatoires aigües, persistantes ou chroniques et peut évoluer vers une forme fulminante,
une cirrhose ou un cancer hépatique. Le modèle de l’hépatite murine induite par le virus de l’hépatite
murine de type 3 (MHV3) permet d’étudier et de mieux comprendre les causes d’une réponse
inflammatoire anormale et les mécanismes immunitaires mis en œuvre pour éliminer le virus.
Suite à l’infection virale, la tolérance immunitaire hépatique est rompue. Les mécanismes de
l’immunité innée et de l’immunité spécifique sont alors activés pour contrôler l’élimination virale.
L’infection hépatique provoque rapidement le recrutement et l’activation de cellules associées à la
réponse immunitaire innée, les cellules dendritiques, les cellules endothéliales sinusoïdales, les
macrophages, les cellules NK, et les neutrophiles, ainsi que la sécrétion de chémokines et de cytokines
pro-inflammatoires, telles que l’IL6, le TNF-α et l’IFN-γ (Jacques, 2008). La rupture de l’état de tolérance
hépatique accompagnée de la réponse inflammatoire permet alors l’activation des cellules de l’immunité
spécifique ou adaptative, les lymphocytes. La réponse immunitaire spécifique développée par le sujet
infecté doit être adaptée au type d’infection auquel il fait face, afin de contrôler efficacement
l’élimination du virus et lui conférer une résistance à l’infection. Ainsi, les souris infectées par le virus
MHV3 développant une réponse immunitaire spécifique de type Th1 sont résistantes, en revanche les
souris développant une réponse Th2 sont plus susceptibles à l’infection et présentent de forts taux de
mortalité (Pope et al., 1996).
Actuellement, de nombreuses études sont menées afin de comprendre les causes des désordres
inflammatoires provoqués lors de l’hépatite virale, ainsi que les interactions entre les différents
mécanismes de l’immunité innée et spécifique. L’un des modèles viraux les plus utilisés pour étudier ces
mécanismes est le virus de l’hépatite murine MHV3. Nous verrons donc à travers cette synthèse
bibliographique, les différentes avancées réalisées en matière de connaissance des mécanismes
immunitaires de lutte contre l’infection hépatique par le virus MHV3.
Cette synthèse présentera tout d’abord le virus de l’hépatite murine MHV3, ces cellules cibles,
différents variants de pathogénicité, ainsi que les récepteurs spécifiques et non spécifiques du virus
MHV3. Nous présenterons ensuite l’ensemble des mécanismes immunitaires, actuellement connus,
développés en réponse à l’infection par le virus MHV3. Les mécanismes de l’immunité innée impliquent
les cellules présentatrices d’antigènes, les macrophages, les cellules tueuses naturelles (NK) et les
neutrophiles et forment la réponse inflammatoire. Les mécanismes de l’immunité spécifique sollicités
suite à la réponse inflammatoire sont variables et impliquent les lymphocytes T tueurs naturels (NKT) qui
sont également impliqués dans l’immunité innée, les lymphocytes T régulateurs FoxP3+ CD4+ CD25+, les
lymphocytes T CD4+ et T CD8+, les lymphocytes T doubles négatifs CD4- CD8- et les lymphocytes B.

M.C. Manus / Les mécanismes immunitaires de lutte contre l’infection hépatique par le virus MHV3 ©
4
I. Le virus de l’hépatite murine de type 3 : MHV3
A. Le virus MHV3
Le virus de l’hépatite murine de type 3 MHV3 fait partie de la famille des coronavirus,
Coronaviridiae. Le sérotype MHV3 est le plus virulent de toutes les souches de virus de l’hépatite murine.
Il induit des dommages tissulaires au niveau de nombreux organes, touchant d’abord le foie, puis les
organes lymphoïdes, la rate, le thymus et le système nerveux central (Jacques, 2008 ; Zhou et al., 2010 ;
Chen et al., 2011). Au niveau hépatique, le virus MHV3 peut induire une hépatite aigüe ou chronique en
fonction de la génétique de l’hôte (résistance ou non), de son âge et de l’état de son système
immunitaire (Jacques et al., 2009a). La gravité de l’hépatite dépend également de l’intensité de la
réponse inflammatoire mise en œuvre par le sujet infecté (Jacques et al., 2008).
B. Infection hépatique : Cellules cibles et variants de pathogénicité
Lors de l’infection hépatique, le virus MHV3 peut se répliquer dans les cellules endothéliales
sinusoïdales (LSEC), les cellules de Kupffer (KC), les cellules tueuses naturelles (NK) et les cellules T
tueuses naturelles (NKT) et les hépatocytes qui sont les cellules cibles finales des virus de l’hépatite
murine (Martin et al., 1994 ; Jacques et al., 2008). Il s’en suit une mort cellulaire, une immunodéficience
dans différents organes lymphoïdes, une nécrose hépatique (annexe 1), et une hépatite fulminante
conduisant à la mort des souris C57BL/6 témoins, 3 à 5 jours après infection (Martin et al., 1994).
Différents variants de pathogénicité sont utilisés pour mieux comprendre les conséquences de
l’infection hépatique par le virus MHV3 (Tableau I). Le virus pathogène L2-MHV3 présente un tropisme
cellulaire KC+, LSEC+ et peut donc infecter les cellules de Kupffer et les cellules endothéliales
sinusoïdales du foie. Le virus moyennement atténué 51.6-MHV3 peut infecter uniquement les cellules de
Kupffer, tropisme KC+,LSEC-. Le virus atténué CL12-MHV3 ne peut infecter ni les cellules de Kupffer, ni
les cellules endothéliales sinusoïdales, son tropisme est KC-/LSEC-. Le virus YAC-MHV3 est un virus non
pathogène (Jacques et al., 2008).
Tableau I. Caractéristiques de pathogénicité et de tropisme cellulaire hépatique de variants du virus de
l’hépatite murine de type 3 (MV3) observées chez des souris témoins C57BL/6 (type sauvage) (D’après
Jacques et al., 2008).
L2-MHV3 51.6-MHV3 CL12-MHV3 YAC-MHV3
Infection des KC Oui Oui Non Faible réplication
Infection des LSEC Oui Non Non Non
Histopathologie Hépatite aigüe
fulminante avec
nécrose totale du
foie
Hépatite aigüe avec
nécrose intra
hépatique
Hépatite légère avec
des foyers
inflammatoires
périvasculaires
Hépatite sub-
clinique avec des
foyers
inflammatoires
locaux
Immunodéficience Oui (3 jours a.i) Oui Non Non
Mortalité (jours a.i.) ≈ 3-4 ≈ 5-9 ≈ 8-10 Elimination du virus
KC : Cellules de Kupffer, LSEC : Cellules endothéliales sinusoïdales, a.i : après infection
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
1
/
20
100%