Lire l'article complet

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XVI - n° 9 - novembre 2012
258
Échos des congrès
© Thomas Roske
48e Congrès de l’EASD
Une étape de réflexion concernant
le traitement du diabète de type 1
Berlin, du 1er au 5 octobre 2012
48th EASD Meeting: a checkpoint for the treatment of type 1 diabetes
Bertrand Duvillié*
* Faculté Necker, Paris.
Le congrès de la Société européenne pour l’étude du diabète (EASD) s’est déroulé
cette année à Berlin en Allemagne et a réuni 18 000 participants. Voici 12 ans
maintenant que les premiers essais cliniques de greffes d’îlots ont été effectués
à Edmonton sur des patients atteints de diabète de type 1, et l’heure est venue
de dresser le bilan des risques encourus et des améliorations apportées par cette
technique. C’est également le moment de faire le point sur les modèles expérimen-
taux et de s’interroger sur l’extrapolation des modèles de rongeurs à l’homme.
Diabète de type 1
D. Quintin (Lille) [1] a présenté une étude clinique qui
analyse l’évolution des complications du diabète chez
des patients ayant reçu une greffe d’îlots depuis 5 ans.
Les auteurs ont montré que la transplantation d’îlots
dans un groupe de 21 patients n’était pas associée
à des complications cardiovasculaires sévères, bien
que certains soient traités pour une coronaropathie
(angioplastie avec pose d’un stent). La fonction rénale
et l’acuité visuelle sont restées stables. De façon intéres-
sante, la vitesse de transmission nerveuse sensitive est
améliorée. L’amélioration neurologique est corrélée
avec le peptide C et la glycémie, avec le β-score pour les
paramètres de sensibilité, avec l’hémoglobine glyquée
et les triglycérides pour l’amplitude de la réponse ner-
veuse. L’ensemble de ces données suggère que, malgré
le régime immunosuppresseur, le protocole d’Edmon-
ton est capable de stabiliser et même d’améliorer les
complications du diabète (essai clinique NCT01123187).
Le développement des nouvelles stratégies qui visent
à traiter le diabète de type 1 chez l’homme repose sur
des études réalisées chez les rongeurs. Il est donc
important de savoir dans quelle mesure ces modèles
expérimentaux sont représentatifs du diabète chez
l’homme. A. Jörns et al. (Hanovre, Allemagne) [2] ont
comparé l’infiltrat de cellules immunitaires ainsi que le
profil d’expression des cytokines dans des îlots prove-
nant de pancréas des 4 types de modèles expérimen-
taux utilisés en laboratoire – à savoir : la souris NOD
(non-obese diabetic), le rat BB (bio-breeding rat), le rat
LEW.1AR1-iddm, et le rat Komeda – avec les îlots de
patients diabétiques de type 1. Ils ont ainsi montré
que les principales cellules du système immunitaire, les
lymphocytes T cytotoxiques CD8+, les lymphocytes T
CD4+ et les macrophages CD68, sont similaires chez
tous ces animaux et chez l’homme. Les cytokines dont le
Tumor Necrosis Factor α (TNFα), l’interleukine 1β (IL-1β)
et l’interféron γ (IFNγ) étaient exprimés dans tous les
groupes au niveau de l’infiltrat de cellules immunitaires,
conduisant à l’apoptose des cellules β. L’exception est le
rat Komeda, pour lequel on observe une prédominance
de l’IFNγ par rapport à l’IL-1β. Ces modèles animaux
semblent donc représentatifs du diabète humain et
nous fournissent des éléments de compréhension
importants pour envisager de nouvelles stratégies
thérapeutiques.
Le laboratoire de F. Bosch (Barcelone, Espagne) s’est
penché sur le rôle protecteur du facteur Insulin-like
Growth Factor 1 (IGF-1) vis-à-vis de la réponse auto-

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XVI - n° 9 - novembre 2012
259
Une étape de réflexion concernant le traitement du diabète de type 1
immune (3). Pour réaliser cette étude, son laboratoire
a produit des souris NOD transgéniques qui expri-
ment le facteur IGF-1 sous le contrôle du promoteur
de l’insuline (RIP). Elles ont été comparées à des souris
NOD sauvages (témoins). Environ 60 % des animaux
témoins ont développé un diabète à l’âge de 8 mois,
alors que seulement 10 % des souris NOD RIP-IGF-1
étaient diabétiques à l’âge de 10 mois. Une analyse
immunohistochimique avec des anticorps anti-insuline
a révélé que les cellules β étaient préservées chez les
souris RIP-IGF-1. De plus, l’infiltration de leucocytes
chez les souris transgéniques était plus faible que chez
les témoins. Cette étude indique donc que l’expression
locale d’IGF-1 protège des souris prédiposées au dia-
bète contre l’infiltration des cellules immunitaires et la
mort des cellules β. Cet effet semble prometteur, mais
il faudra néanmoins rester prudent, car l’IGF-1 pourrait
favoriser la survenue de tumeurs.
Mitochondries et diabète de type2
On sait que les mitochondries jouent un rôle très
important dans la sécrétion d’insuline en fournissant
l’énergie nécessaire à la cellule β pour assurer ce tra-
vail. Le congrès de l’EASD a été l’occasion de rapporter
des travaux qui mettent en évidence l’importance de
plusieurs facteurs mitochondriaux.
Le facteur TB1FM
V. Sharoyko et al. (Malmö, Suède) [4] ont étudié le fac-
teur de transcription et de traduction mitochondrial B1
(TFB1M). Une étude génétique d’association avait en
effet montré que TFB1M pourrait être impliqué dans le
diabète de type 2. Sharoyko et al. ont produit et ana-
lysé des souris qui présentent une délétion de TFB1M
spécifiquement dans la cellule β. Les îlots de ces souris
sécrétaient moins d’insuline en réponse au glucose que
les îlots témoins. Ces îlots TFB1M−/− consommaient
moins d’oxygène en réponse au glucose et contenaient
moins d’ATP que les îlots sauvages (WT). La densité
des granules de sécrétion d’insuline était réduite de
moitié chez les cellules β des mutants par rapport aux
témoins. La surface des mitochondries passait de 7 %,
chez les témoins, à 19 % dans les cellules β TFB1M−/−.
Enfin, 4 mois après la naissance, les souris TFB1M−/−
développaient un diabète avec une infiltration de
macrophages dans les îlots. L’ensemble de ces résultats
démontre que TFB1M joue un rôle important dans la
fonction des cellules β. L’hypothèse principale est que
ce facteur pourrait affecter la synthèse des protéines
mitochondriales, conduisant à un diabète de type 2. Le
fait que TFB1M ait été identifié chez l’homme comme
un facteur de risque du diabète de type 2 joue en faveur
de cette hypothèse.
Fission des mitochondries
et sécrétion d’insuline
Les mitochondries appartiennent à un réseau dyna-
mique où se produisent continuellement des événe-
ments de fusion/fission. La fusion est contrôlée par les
mitofusines 1 et 2, et la protéine d’atrophie optique
OPA1. La fission des mitochondries est contrôlée par
la protéine de fission 1 (Fis1) et la protéine apparen-
tée à la dynamine (Drp1). J. Schultz et al. (Rostock,
Allemagne) [5] ont examiné l’hypothèse d’un rôle de
Fis1 dans la sécrétion d’insuline. Pour cela, ils ont uti-
lisé des lignées de cellules β Ins1, qu’ils ont infectées
avec des lentivirus exprimant un short hairpin RNA
(shRNA) de Fis1. En conséquence, un allongement et
un regroupement des mitochondries étaient observés.
De plus, la sécrétion d’insuline en réponse au glucose
était altérée dans les cellules β ayant une réduction de
l’expression de Fis1 (figure). Ces données suggèrent
que la structure de la mitochondrie est un point impor-
tant pour sa fonction. Le facteur Fis1 paraît crucial
pour ce processus. Ce facteur pourrait représenter à
l’avenir un sujet d’étude pour le traitement du diabète
de type 2.
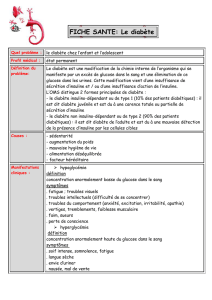
Figure. Le facteur mitochondrial Fis1 contrôle la sécrétion d’insuline. Des cellules Ins1 ont été
infectées avec un lentivirus portant un shRNA Fis1, puis analysées pour leur capacité de sécrétion
d’insuline en réponse au glucose.
2,8Glucose (mM)
Contrôle
20
50
100
2,8 20
Sécrétion d’insuline
shFis1

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XVI - n° 9 - novembre 2012
260
Échos des congrès
FRDBT00147 - Novembre 2010 - © Lilly - Tous droits de reproduction réservés. Lilly France 13, rue Pagès - 92 158 Suresnes Cedex - Tel : 01 55 49 34 34 - Fax : 01 41 44 02 47 - www.lilly.fr - S.A.S. au capital de 358 511 701 e - 609 849 153 R.C.S. Nanterre
Votre vie, notre inspiration.
Lilly Diabète
17050_AP_diabete_A4_E2.indd 1 28/02/11 14:56
Récepteur à la sérotonine HTb2 et sécrétion
d’insuline : le rôle des mitochondries
La sérotonine, encore appelée 5-hydroxytryptamine
(5-HT), est une monoamine servant de neurotrans-
metteur dans le système nerveux central. La séroto-
nine est essentielle dans l’excitation et l’inhibition des
neurones, la motilité gastro-intestinale, la contraction
des muscles lisses et la contraction et la dilatation
vasculaires. En dehors de son activité sur le système
nerveux central, la sérotonine est exprimée dans le
pancréas endocrine. Notamment, on a émis l’hypo-
thèse qu’elle module la sécrétion d’insuline. Toutefois,
ces données sont controversées. Parmi les différentes
formes de récepteurs de la sérotonine, 5-HT1a, 5-HT1d,
5-HT2a sont détectées à la fois dans les cellules α et β.
La forme 5-HT2b ne semble présente, chez l’homme
et les rongeurs, que dans les cellules β. La stimulation
du récepteur 5-HT2b par un agoniste (α-MET) aug-
mente la sécrétion d’insuline en réponse au glucose. En
revanche, l’extinction de l’expression du gène 5-HT2b
produite par un small interfering RNA (siRNA) réduit la
sécrétion d’insuline en réponse au glucose de 30 %
dans des cellules INS(832/13) [6]. Enfin, l’activation du
récepteur 5-HT2b augmente la respiration mitochon-
driale. Ces données suggèrent donc fortement que la
signalisation par le récepteur 5-HT2b est couplée à la
respiration mitochondriale.
MicroARN et cellules β
La gestation et l’obésité sont associées à une insulino-
résistance et une demande d’insuline accrue. Dans
ces situations, une augmentation de la masse de cel-
lules β est nécessaire afin de maintenir la glycémie. Les
mécanismes cellulaires et moléculaires qui permettent
cette adaptation sont encore très largement discutés
à ce jour. C. Jacovetti et al. (Lausanne, Suisse) [7] ont
proposé l’hypothèse d’une régulation de la masse de
cellules β par les microARN. Cette famille de molécules
est constituée d’ARN non codants, qui jouent un rôle très
important dans l’expression des gènes, en se liant sur la
partie 3’-UTR de leurs ARN cibles. Ainsi, ils inhibent par-
tiellement leur transcription ou leur stabilité. Jacovetti
et al. ont examiné le profil d’expression des microARN
dans des modèles dans lesquels on observe une com-
pensation de la masse de cellules β : rates gestantes,
souris exposées à un régime riche en graisse, ou souris
obèses db/db de 6 semaines. Les auteurs ont observé
une réduction de l’expression de miR-338-3p dans les
îlots de ces différents modèles, par rapport à un groupe
témoin. La réduction de miR-338-3p est également
observée dans des îlots de rats ou des îlots humains
traités par l’estradiol ou l’exendine 4. Cette réduction
conduisait à une augmentation de la prolifération
et de la survie des cellules β exposées à des stimuli
proapoptotiques. Enfin, la réduction de l’expression de
miR-338-3p à l’aide d’un antisens induisait une diminu-
tion de l’expression de gènes proapoptotiques et une
augmentation de l’expression des gènes antiapopto-
tiques et de prolifération dans des cellules β de rats
ou humaines. Ces résultats montrent l’implication de
miR-338-3p dans l’adaptation cellulaire à la gestation,
mais fournissent également de nouveaux outils sus-
ceptibles d’être utilisés pour induire une régénération
des cellules β dans des modèles pathologiques.
■
Les références sont consultables dans :
Diabetologia.
Clinical and Experimental Diabetes and Metabolism. Journal
of the European Association for the Study of Diabetes (EASD)
2012;55(suppl.1).
1.
Quintin D et al. Evolution of diabetes complications 5 years
after islet transplantation (IT) with the Edmonton immuno-
suppressive regimen. P434.
2.
Jörns A et al. The pathology underlying beta cell destruction
in four type 1 diabetes animal models in comparison with the
human situation. P446.
3.Mallol C et al. Pancreatic overexpression of IGF-I protects
NOD mice from autoimmune diabete. P444.
4.Sharoyko V et al. Deficiency of TFB1M leads to beta cell
dysfonction and development of diabetes. OP17.
5.Schultz J et al. Fis1, a key regulator of the mitochondrial
network, controls glucose responsivness in beta cells. OP17.
6.Bennet H et al. Activation of the 5-HT2b receptor in INS-1
cells couples to mitochondrial respiration and potentiates
glucose insulin stimulated secretion. OP17.
7.Jacovetti C et al. Role of the microRNA miR-338-3p in com-
pensatory beta-cell mass expansion during pregnancy and
obesity. OP 36.
Références
1
/
3
100%