R e v u e d e ... Histoire de Shadoks

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XVII - n° 6 - juin 2013
162162
Revue de presse
Coordination : Estelle Louiset (Rouen)
Histoire de Shadoks
Polémique sur les eff ets
des traitements par
hormone de croissance
Sécrétion de mélatonine
et risque de diabète
Histoire de Shadoks
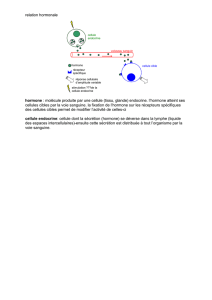
L’hyperaldostéronisme primaire est dû, dans la moitié
des cas, à un adénome de Conn et, pour l’autre moitié,
à une hyperplasie de la zone glomérulée touchant l’une
des glandes surrénales ou les 2. Les études moléculaires
récentes ont révélé que plus de 1/3 des cas familiaux
et sporadiques d’hyperaldostéronisme primaire sont
associés à des mutations germinales et somatiques du
gène codant pour le canal potassique KCNJ5. Le réseau
de recherche européen de l’Institut national du cancer
(Inca), en collaboration avec le réseau français Cortico-
médullo tumeurs endocrines (COMETE), rapporte pour
la première fois des mutations somatiques des gènes
codant pour les pompes Na+/K+ ATPase (ATP1A1) et
Ca
2+
ATPase (ATP2B3) dans des adénomes de Conn. En
permettant l’effl ux de 3 Na+ en contrepartie d’un infl ux
de 2 K+, la pompe Na+/K+ ATPase maintient le potentiel
de repos des cellules. En extrayant des ions Ca
2+
des
cellules, la pompe Ca
2+
ATPase participe au contrôle
de l’homéostasie calcique. Le séquençage des 2 gènes,
réalisé sur plus de 300 tissus d’adénomes de Conn,
a révélé la présence de mutations somatiques chez
21 sujets (6,8 %). Dans le gène ATP1A1, des mutations
ponctuelles (c.311T>G, c.995T>G et c.1738A>G) entraî-
nant les substitutions Leu104Arg, Val332Gly et Ile580Val,
ainsi qu’une délétion (c.299_313delTCTCAATGTTACTGT)
responsable d’une perte de la région Phe100_Leu104,
ont été détectées. Dans le gène ATP2B3 des délétions
(c.1272_1277delGCTGGT, c.1273_1278delCTGGTC
et c.1277_1282delTCGTGG) à l’origine de la perte
de 2 acides aminés 425-426 ou 426-427 (p.Leu425_
Val426del ou p.Val426_Val427del) ont été observées.
Les études fonctionnelles réalisées in vitro sur des cel-
lules montrent que les mutations induisent une perte
de l’activité enzymatique des pompes. Les mutations
du gène ATP1A1 engendrent une dépolarisation des
cellules tumorales qui pourrait faciliter l’ouverture des
canaux calciques voltage-dépendants permettant un
infl ux de calcium. L’augmentation du calcium cytoso-
lique stimulerait à son tour la production d’aldostérone.
De même, les mutations d’ATP2B3 à l’origine d’une
perte de fonction de la pompe Ca2+ ATPase pourraient
induire une élévation de la concentration cellulaire de
Ca2+ capable d’augmenter la synthèse d’aldostérone.
Il est à noter qu’aucune mutation germinale des gènes
ATP1A1 et ATP2B3 n’a été détectée, que ce soit chez ces
quelque 300 patients atteints d’un adénome de Conn
ou chez les 18 cas d’hyperaldostéronisme familial et
91 cas d’hyperplasie bilatérale associée à un hyperal-
dostéronisme primaire. La sécrétion d’aldostérone par
les cellules corticosurrénaliennes est donc contrôlée par
les Na
+
/K
+
ATPase et Ca
2+
ATPase, qui, tels des Shadoks,
pompaient, pompaient, pompaient…
E. Louiset (Rouen)
• Beuschlein F et al. Nat Genet 2013;45:440-4.
Polémique sur les eff ets des traitements
par hormone de croissance
Après la polémique sur les eff ets cancérigènes poten-
tiels du traitement par hormone de croissance, plu-
sieurs études ont infi rmé que les enfants traités par
hormone de croissance aient un risque accru de déve-
lopper un cancer de novo par rapport à la population
générale. Mais un signal d’alerte a été émis concer-
nant un sous-groupe d’enfants ayant survécu à un
cancer, chez qui un risque de développer une néoplasie
secondaire aurait été observé. Le défi cit en hormone
de croissance est une complication fréquente chez
des enfants ayant survécu à un cancer. La cause de
ce défi cit peut être la tumeur, lorsqu’elle atteint l’axe
hypothalamohypo physaire, ou bien les traitements
instaurés (chimiothérapie et radiothérapie). Le but de
cette étude était d’analyser, de façon rétrospective,
2 cohortes prospectives et d’établir l’incidence de néo-
plasie secondaire chez des enfants ayant survécu à un
cancer. La première base de données, GeNeSIS (Genetics
and Neuroendocrinology of Short Stature International
Study), attachée au groupe pharmaceutique Lilly, col-
lecte des informations sur la prise en charge et le suivi
des enfants ayant un retard de croissance. La deuxième,
HypoCCS (Hypopituitary Control and Complications
Study), est un programme de veille sanitaire chez des
adultes traités par hormone de croissance. Les patients
ayant eu un cancer avant l’âge de 21 ans ont été inclus
dans l’étude, soit 421 (394 traités par hormone de crois-
sance et 27 non traités) dans la base GeNeSIS, et 280
(252 traités et 28 non traités) dans la base HypoCCS.
Le cancer secondaire était défi ni par la présence d’un
cancer bénin ou malin apparu après le cancer primi-
tif, en excluant les métastases et les rechutes du can-
cer primitif. Dans GeNeSIS, l’incidence de néoplasie
secondaire était de 3,8 % chez les enfants traités par
hormone de croissance (IC95 : 2,2-6,2), alors qu’il n’y a eu
aucun cas rapporté chez les sujets non traités. Dans la
base HypoCSS, l’incidence était de 6,0 % (IC95 : 3,4-9,6)
chez les patients traités par hormone de croissance, et
de 7,1 % (IC95 : 0,9-23,5) chez les patients non traités.
>>>

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XVII - n° 6 - juin 2013
164164
Revue de presse
AVIS AUX LECTEURS
Les revues Edimark sont publiées en toute indépendance et sous l’unique et entière responsabilité du directeur de la publication et
du rédacteur en chef.
Le comité de rédaction est composé d’une dizaine de praticiens (chercheurs, hospitaliers, universitaires et libéraux), installés partout
en France, qui représentent, dans leur diversité (lieu et mode d’exercice, domaine de prédilection, âge, etc.), la pluralité de la discipline.
L’équipe se réunit 2 ou 3 fois par an pour débattre des sujets et des auteurs à publier.
La qualité des textes est garantie par la sollicitation systématique d’une relecture scientifi que en double aveugle, l’implication d’un
service de rédaction/révision in situ et la validation des épreuves par les auteurs et les rédacteurs en chef.
Notre publication répond aux critères d’exigence de la presse :
· accréditation par la CPPAP (Commission paritaire des publications et agences de presse) réservée aux revues sur abonnements,
· adhésion au SPEPS (Syndicat de la presse et de l’édition des professions de santé),
· indexation dans la base de données INIST-CNRS et Thomson Reuters et partenariats avec les sociétés savantes (SFE, …),
· déclaration publique de lien d’intérêts demandée à nos auteurs,
· identifi cation claire et transparente des espaces publicitaires et des publirédactionnels en marge des articles scientifi ques.
Si les auteurs concluent que leurs résultats
sont en accord avec un risque accru de néo-
plasie secondaire à la suite du traitement par
hormone de croissance des enfants ayant
survécu à un cancer, il est bien diffi cile de
passer à côté des faiblesses de l’étude. La
disproportion entre le nombre d’enfants
traités et non traités dans les bases de don-
nées ne permet pas d’analyse statistique
fi able. De plus, il n’y a pas d’information sur
le type de chimiothérapie et les doses de
radiothérapie utilisés dans le traitement du
cancer primitif, composants connus pour
être des facteurs de risque de néoplasies
secondaires et sources de biais potentiels
pour cette étude. Même si une attitude cir-
conspecte reste de mise dans l’instauration
d’un traitement par hormone de croissance
dans ce groupe de patients, des études pros-
pectives devront être réalisées pour éclaircir
l’eff et cancérigène potentiel du traitement
par hormone de croissance chez les enfants
ayant survécu à un cancer.
A. Naccache (Rouen)
• Woodmansee WW et al. European Journal of Endocrinology.
2013;168:565-7.
Sécrétion de mélatonine et risque
de diabète
La mélatonine est une hormone sécrétée par
la glande pinéale sous le contrôle de l’hor-
loge biologique en fonction de la lumière.
Sa sécrétion suit un rythme circadien, avec
un pic entre 3 et 5 h après l’endormissement.
Les récepteurs de la mélatonine sont pré-
sents dans les cellules β pancréatiques. Des
mutations du récepteur ont été associées à
un risque accru d’insulinorésistance et de
diabète de type 2. L’étude de McMullan et
al. avait pour objectif d’évaluer le lien entre
la sécrétion de mélatonine et le risque de
développer un diabète de type 2. Pour cela,
les auteurs ont étudié, au sein de la cohorte
de la Nurses’ Health Study, 370 femmes
non diabétiques à l’inclusion en 2000 qui
ont développé par la suite un diabète et
370 femmes n’ayant pas développé de dia-
bète. Les participantes ont été appariées sur
de nombreuses caractéristiques cliniques
et d’habitudes de vie, comme la qualité du
sommeil. La sécrétion de mélatonine a été
évaluée par le rapport de la 6-sulfatoxymé-
latonine (6-S-MT) sur la créatinine urinaire
(créat.). Les médianes étaient de 28,2 ng/mg
chez les patientes et 36,3 ng/mg chez les
témoins. La sécrétion de mélatonine variait
considérablement parmi les participantes.
Comparativement aux femmes dans la
catégorie la plus haute du rapport 6-S-MT/
créat. (67,0 ng/mg), celles de la catégorie la
plus basse (14,4 ng/mg) avaient un risque
multiplié par 2 de développer un diabète
de type 2 (OR en analyse multivariée de 2,17
[IC95 : 1,18-3,98]). Cela se traduit par un écart
absolu de l’incidence du diabète de 5 cas
pour 1 000 personnes par année. Cette ana-
lyse montre donc qu’une faible sécrétion de
mélatonine est associée à un risque accru
de diabète de type 2. Des études d’obser-
vation ont révélé que le travail nocturne et
la restriction de sommeil sont associés au
diabète. Il reste à déterminer si l’augmenta-
tion des niveaux de mélatonine (endogène
via un sommeil prolongé ou exogène via une
supplémentation en mélatonine) peut aug-
menter la sensibilité à l’insuline et diminuer
l’incidence du diabète de type 2.
C. Sanz (Toulouse)
• McMullan CJ et al. JAMA 2013;309(13):1388-96.
>>>
Les auteurs n’ont pas précisé leurs éventuels liens d’intérêts.
1
/
2
100%