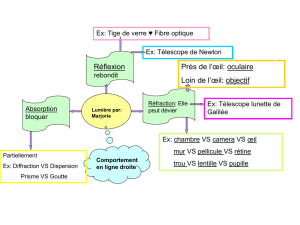
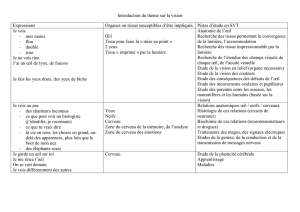
VII-3 : DYSFONCTIONNEMENTS VISUELS ASSOCIÉS - Lodel

Œil et Physiologie de la Vision - VII-3. 2ème partie
VII-3 : DYSFONCTIONNEMENTS VISUELS ASSOCIÉS À
QUELQUES MALADIES PÉDIATRIQUES MÉTABOLIQUES,
SYSTÉMIQUES et NEUROLOGIQUES. Apport du bilan
électrophysiologique
Deuxième partie
Florence Rigaudière
Eliane Delouvrier
Pour citer ce document
Florence Rigaudière et Eliane Delouvrier, «VII-3 : DYSFONCTIONNEMENTS VISUELS
ASSOCIÉS À QUELQUES MALADIES PÉDIATRIQUES MÉTABOLIQUES, SYSTÉMIQUES et
NEUROLOGIQUES. Apport du bilan électrophysiologique», Oeil et physiologie de la vision
[En ligne], VII-Electrophysiologie pédiatrique, mis à jour le 18/06/2013, URL :
http://lodel.irevues.inist.fr/oeiletphysiologiedelavision/index.php?id=241,
doi:10.4267/oeiletphysiologiedelavision.241
Plan
Erreurs du métabolisme lysosomal
Plusieurs types de pathologies lysosomales
Manifestations des pathologies lysosomales
Mucopolysaccharidoses
Classification
Physiopathologie
Signes cliniques
Dysfonctionnements visuels
Mucopolysaccharidose de type I S (Scheie) : exemple
Gangliosidoses à GM2 : groupe des sphingolipidoses
Classification : Tay-Sachs, Sandhoff, variant AB
Physiopathologie
Génétique
Signes cliniques
Traitement
Gangliosidose à GM2. Exemple : maladie de Tay-Sachs
Céroïdes lipofuscinoses neuronales
Classification
Physiopathologie
Génétique
Signes cliniques. Rétinopathie et atrophie optique
Traitement
Céroïde lipofuscinose. Exemples : CLN2, CLN3, CLN8
Synthèse : fonctionnements visuels et maladies lysosomales
Erreurs de la glycosylation
Anomalies de la glycosylation : CDG
1

Œil et Physiologie de la Vision - VII-3. 2ème partie
PMM2-CDG (CDG-Ia)
PMM2-CDG (CDG-Ia) révélé par des troubles oculomoteurs & un retard de maturation : exemple
Apraxie oculomotrice et CDG
Commentaires
DPM1-CDG (CDG-Ie)
DPM1-CDG (CDG-Ie) : exemple
Synthèse du fonctionnement visuel des deux patients présentés, atteints l’un de CDG-Ia et l’autre de CDG-Ie
Erreurs du métabolisme peroxysomal
Fonction des peroxysomes
Déficits des fonctions peroxysomales
Maladie de Refsum infantile
Maladie de Refsum infantile : exemple
Commentaire
Adrénoleucodystrophie liée à l’X
Adrénoleucodystrophie liée à l’X : exemple
2

Œil et Physiologie de la Vision - VII-3. 2ème partie
Texte intégral
Collaboration : Anne Jacob & David Lebrun pour la prise en charge des enfants et
enregistrements des bilans.
Erreurs du métabolisme lysosomal
Les lysosomes sont des organites intracellulaires présents dans toutes les cellules sauf
les hématies. Ils sont responsables de la digestion (dégradation) intracellulaire de
particules phagocytées ou de structures sénescentes. Les enzymes cataboliques qu’ils
contiennent sont des glycosidases, lipases, protéases et nucléases.
Les maladies lysosomales, encore appelées maladies de surcharge, sont dues à des
anomalies génétiques de ces enzymes, entraînant l’accumulation de substrats
incomplètement métabolisés dans ces organites d’où leur augmentation de taille ou
« ballonisation » et dysfonctionnement de la cellule. Il s’ensuit une organomégalie, des
anomalies du tissu conjonctif, du cartilage, des os, une atteinte oculaire et du système
nerveux (Zschocke J. & Hoffmann GF., 2005).
Christian de Duve –médecin et chimiste belge- les a découverts en 1955 ; pour l’ensemble de ses découvertes,
et travaux, il a partagé, avec Albert Claude et George Emil Palade, le prix Nobel de médecine en 1974. Note
due à J Ph Onolfo.
Plusieurs types de pathologies lysosomales
Traditionnellement les maladies lysosomales étaient classées en plusieurs catégories
selon le type de substrat accumulé (Zschocke J. & Hoffmann GF., 2005) (p.116-117) et
recouvraient seulement les déficits en hydrolases lysosomales.
La classification a été actualisée ; le concept de maladie lysosomale a été élargi pour
inclure d’autres mécanismes physiopathologiques : déficits en protéines responsables du
trafic entre le lysosome et les autres compartiments intracellulaires, déficits de protéines
chaperonnes des enzymes lysosomales, déficits en protéines lysosomales non
enzymatiques, déficits du transport lysosomal (De Lonlay P. et al., 2013).
Manifestations des pathologies lysosomales
De nombreuses mutations génétiques sont à l’origine de plus de 45 maladies lysosomales
(Urbanelli L. et al., 2011).
Symptomatologie progressive
Ces maladies sont le plus souvent évoquées devant une symptomatologie progressive
avec dysmorphie évolutive, anomalies osseuses, cardiaques, hépatosplénomégalie, une
atteinte neurologique avec retard de développement et régression psychomotrice,
épilepsie, hypotonie, ataxie, spasticité, hyperexcitabilité (Defoort-Dhellemmes S. et al.,
2012). Elles peuvent être de présentation précoce, voire in utero, ou bien se manifester
tardivement chez l’adulte.
Atteintes visuelles
Les atteintes visuelles sont rarement au premier plan mais peuvent venir en grever
l’évolution. Elles peuvent toucher la cornée, le cristallin, la rétine et/ou donner des
neuropathies optiques (résumé figure VII-3-35, d’après (Menache CC., Haenggeli Ch-A. &
Safran AB., 2004), (Ashworth, J.L. et al., 2006a)).
3

Œil et Physiologie de la Vision - VII-3. 2ème partie
Traitement
Dans la majorité des cas, le traitement est symptomatique. Un bénéfice est apporté à
certaines manifestations par transplantation de moelle (Ohlemiller K.K. et al., 2000),
enzymothérapie substitutive (Hoffmann & Mayatepek, 2005), (Beck, 2007) ou utilisation
de molécules activateurs de l’enzyme lysosomal résiduel (Urbanelli et al., 2011).
Ces traitements sont d’autant plus efficaces qu’ils sont administrés tôt, soulignant
l’importance d’un diagnostic précoce ; ils soulèvent d’immenses espoirs. Des recherches
sont en cours (Urbanelli et al., 2011)
Exemples
Pour l’ensemble des pathologies lysosomales, le lecteur intéressé peut se rapporter à
l’ouvrage de Saudubray et coll. 5ième édition 2012 (Saudubray JM, Van den Berghe G &
Walters JH., 2012) (chapitres 39-40).
Nous ne donnons ici que quelques exemples de résultats enregistrés chez des patients
souffrant de Mucopolysaccharidoses, Sphingolipidoses et Céroïdes lipofuscinoses pour
lesquels une exploration fonctionnelle visuelle nous a été demandée en complément du
bilan général et ophtalmologique, à la recherche d’atteintes rétiniennes ou des voies
visuelles.
Mucopolysaccharidoses
Ce sont des anomalies de dégradation des mucopolysaccharides ou glycosaminoglycanes
(GAG) qui sont des sucres-aminés-sulfatés ou acétylés, attachés à un squelette
protéique.
Classification
Une classification en différents types a été établie selon les déficits enzymatiques et les
manifestations systémiques (Ashworth et al., 2006a). Le type I est autosomique
récessif ; il se subdivise en trois : le I H (maladie de Hurler, la plus grave), le I S
(maladie de Scheie, la plus modérée) et le I H/S (maladie de Hurler-Scheie,
intermédiaire). Le type II est lié à l’X. Les autres types III, IV, VI, VII, IX sont
autosomiques récessifs (Ashworth et al., 2006a).
Physiopathologie
Les mucopolysaccharidoses correspondent à de nombreuses maladies héréditaires liées à
un stockage anormal de glycosaminoglycanes non dégradées à l’intérieur des lysosomes
cellulaires, dû à un déficit d’hydrolases acides qui empêchent leur dégradation (Germain,
2002). Des mucopolysaccharides non dégradés s’accumulent dans les tissus, dermatan-
sulfate (tissus conjonctifs), kératane-sulfate (membranes cellulaires), héparane-sulfate
et chondroïtine-sulfate (cartilage, cornées). Ils sont excrétés dans les urines.
Signes cliniques
Ils sont variables selon le type de la mucopolysaccharidose. On retrouve à des degrés
divers, une dysmorphie faciale caractéristique avec traits du visage épaissis,
macrocranie, des déformations progressives du squelette avec dysplasie osseuse et
enraidissement articulaire ainsi qu’une hépatosplénomégalie et une cardiomyopathie.
On peut aussi voir apparaître des signes neurologiques (Hoffmann & Mayatepek, 2005),
un retard psychomoteur progressif (Urbanelli et al., 2011), une surdité. Les hernies
inguinales et ombilicales, les infections des voies aériennes supérieures et respiratoires
sont fréquentes (Zschocke J. & Hoffmann GF., 2005).
4

Œil et Physiologie de la Vision - VII-3. 2ème partie
Le diagnostic se fait essentiellement par l’analyse des mucopolysaccharides dans les
urines et par études enzymatiques (Zschocke J. & Hoffmann GF., 2005)
Dysfonctionnements visuels
Strabisme
Un strabisme est fréquent, plus volontiers une exotropie.
Atteintes cornéennes
Des opacités cornéennes sont présentes chez la plupart des sujets atteints (Ashworth,
J.L. et al., 2006b) de type d’infiltration grisâtre et punctiforme (De Lonlay et al., 2013).
Amétropie
L’hypermétropie et l’astigmatisme sont fréquents en relation avec le changement de
forme de la cornée due à son infiltration par les mucopolysaccharides (Ashworth et al.,
2006a).
Atteinte de l’angle irido-cornéen
L’accumulation des glycosaminoglycanes dans les structures du segment antérieur est à
l’origine d’une obstruction du flux trabéculaire. Il s’en suit une hyperpression intra-
oculaire voire un glaucome (Ashworth et al., 2006a).
Dysfonctionnements rétiniens
La neurorétine peut présenter des pigments avec rétrécissement des vaisseaux et une
atrophie de l'épithélium pigmentaire.
Pour de nombreuses mucopolysaccharidoses, l’accumulation de glycosaminoglycanes se
fait initialement dans les lysosomes des cellules de l'épithélium pigmentaire. Le
dysfonctionnement des neurorétines serait secondaire, avec dégénérescence progressive
des photorécepteurs par un mécanisme encore à préciser (Ohlemiller et al., 2000),
(Hennig A.K. et al., 2004).
° Résultats de l’ERG. Un dysfonctionnement neurorétinien est mis en évidence par des
diminutions d’amplitudes de l’ERG flash (Ohlemiller et al., 2000), (Hennig et al., 2004)
avec une atteinte prépondérante du système scotopique (Ashworth et al., 2006a).
L’ERG serait de type électronégatif uniquement pour le système scotopique pour les
mucopolysaccharidoses de type I H/S (Hurler-Scheie) et pour les deux systèmes
(photopique et scotopique) pour une mucopolysaccharidose de type I H (Hurler) (Tzetzi,
Hamilton, Robinson & Dutton, 2007)).
Ces résultats laissent supposer la présence d’un dysfonctionnement post-réceptoral
portant essentiellement sur la voie ON des bâtonnets pour le premier cas (Hurler-Scheie)
et impliquant les voies ON (cônes et bâtonnets) et OFF (cônes) pour le deuxième cas
(Hurler).
Neuropathie optique
Elle est retrouvée chez 14 % des sujets atteints de différents types de
mucopolysaccharidoses (Ashworth et al., 2006b), mais de façon plus fréquente chez les
patients souffrant de mucopolysaccharidose de type I. Un épaississement du nerf optique
est présent à un stade précoce de la maladie (Schumacher R.G. et al., 2008), avec
parfois un œdème papillaire visible au fond d’œil (Collins, Traboulsi & Maumenee, 1990).
5
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
1
/
45
100%