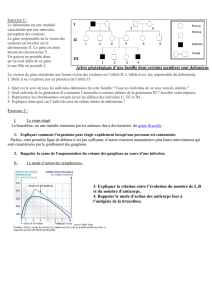
heredite non conventionnelle 21 03 2013 10h

UE : Génétique
!
"#
#$%###$"#
&!!'!#'
'(#)
!!*
*!+(*
'!,)
+(!*!!)+(
#-#'.*(#('/
0!#'#('.*
(1*!+(*
-'''2'.
-''#$32'
-''4/(
('#(1.*
*(
*'#
#'56 7

I/ Hérédité mitochondriale
1- introduction
##('#(8(/'(6!
(!(#!8!8*!*(99(9:;
(!#'8##(8'*)<=8=$5>8#?*('?#'*(
*!'@!8(#'5(6A'B'A5+(
C#5'()*5''!!)+(!!!/'#'#
!#'
)(*((#!*()#')(?''(!8
D'''(4A8''1#**+(8#'+(8!*)+(8
!8(#(')/<#(*#!/#'@!B''5?>E5)
''()-!+(+(/*(#.5'#.((
.(-*'?-*'()*+(!
4(#!#'8!/'#(#+('()*#(
+(./!1-*'!!)E*(F!1###-*'()
'##'**)#(?
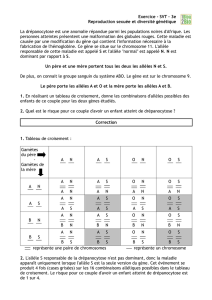
Quelle est la parcularité de ces maladies mitochondriales ?
2- transmission maternelle
'#'('.!!'!#'B!#')8./('
#'+(#'#('*G*'./(8
.5'#'(/!'HI(B'.
E+((.5+((+(.(B*('.(()(!#'/
%#%()1B8J,#(',%8#1##('
!!)8/%()81./'J+(%*
E+(!+(#G#'/'(''#'#8<en gris clair ou 2ème carré et
3ème rond en bas à gauche par exemple>8*!-*5(#(*+(!5J(
()6*1('!''#'(1.
#'56 7

3- L'hétéroplasmie
(#(8(/1B'#'(!'#'
''/#(85A#''@!**)
'#'(!(6#(,#(,/'#
(//#5(#(*'#'(8**)*(/'1KL'
#'(!
**D('('(+(-(6*''8(
6**)<'.(*(B5+((6'#'(!.(>('('(
(8'.6*
(#1ML5(/5B#)''#'
#'(6##!)+('.!!'!#'
–D(6F/5!'*!-*('.(FB*(
F'
#'56 7

II/ L'empreinte génomique
!'*N'(#*'#*'##('(8(#*
(#**8#*!!#*(.6*'?
('O!'?8##*.6*'F?'!E8*(
(#'?8#.*/?/F6*!(+(1*)'.?
8'.((+(1*)'.?*.!#'.
1- Définition
-('?##!
Aenon, l'empreinte génomique n'est pas l'empreinte généque qui elle, est une signature sur le génome,
une carte d'identé généque réalisée grâce à des marqueurs polymorphes dans le domaine de
l'iden#caon humaine.
E*'.**+('?#)B(#'.*
E*'.*+('?#)B(#'.
'H/?*6*8#'/F+(!'.(?
*)#(?(#!*('+(+(?*/'.(/#-8'#+(.'.
/'F'?'.*.+(!*!!)+(
2- Le marquage épigénétique
+(!*!!)+('+(.'(?<*(>8!<(!(
!>(5'HI(BB!#'!*((('/8#'(#('.'/'(
P('*'+(!*!!)+(8*'!-)
(?!-!(?!;
–(?'!!-!(?(!8#)/!
6'.(BJ'.6*((?8*'#!-)'!#!-)'
#)!#*#*/(!-)'!!-)C8!5!
A(/Q!'8#+(!*!!)+(#G.#G((
#((6 !""#"
#'56 07

(#8.*!(?+(.6*1B(#*(
#
'8?(1.*!+(*8'#?*(O(!
'(#('.4(#*'(?6*!
8*(#?8(#*8((B#)#.(#$
*+(+(..-*('#*'#(@8(()(/(#*8
+((1.6*8*'B#)'(? #**!!8
*(**'
%!G-)B#)15''!!)+(!1''.*
!+(*
6*
E*(*B('/'(/#'#'.<B('/#->(
'#*<B('*GN'>-'6**+('
#()+(
–%$+(*/'#('!!)+(R#
(+('.*(/
–*B))'.(I(B(#!!**GN'*N'/#'(*#)'(!
*;
–B))'.(I(B(#!!**GN'
S#%()1#D(((*5)+(/#(5#'
'!/**'()(BI-**')(65-
–&' +(*/'#(*!!)+(#*(+('(
!!!)+('.*((/*#)/)*!'.(/#-/#
'(*#)'(!
6*T?.*#'#!(1.*!+(*
#!)+('?(1*!+(*
(/('#?8-('@!#'!-)?*(6U!!8
!-)'(*#(('(('(!'(
'!/**'.5')(
*B85/(#('('!/**'',#)'!-)8/#'*,
'!-)+('@!*!#,+('#)('.
3- Cycle de l'empreinte au cours de la vie
A!.*(B!!-!(#('('!/**(#(''@!
*!''/
–'(!!? 8.* *'
'@!#)'#(*GN'/#-
–*?B!#')8 .*(*'!*#)!!)'
#8#.1'+(+(!*!!)+(
<!/(',!(#('('!/**#/(*((>
–#('((/8.*('*!##
#'56 V7
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
6
7
8
9
10
11
12
13
14
15
16
17
18
19
1
/
19
100%